namd入门教程(三)
需积分: 29 65 浏览量
2010-11-30
12:05:19
上传
评论
收藏 1.42MB DOC 举报


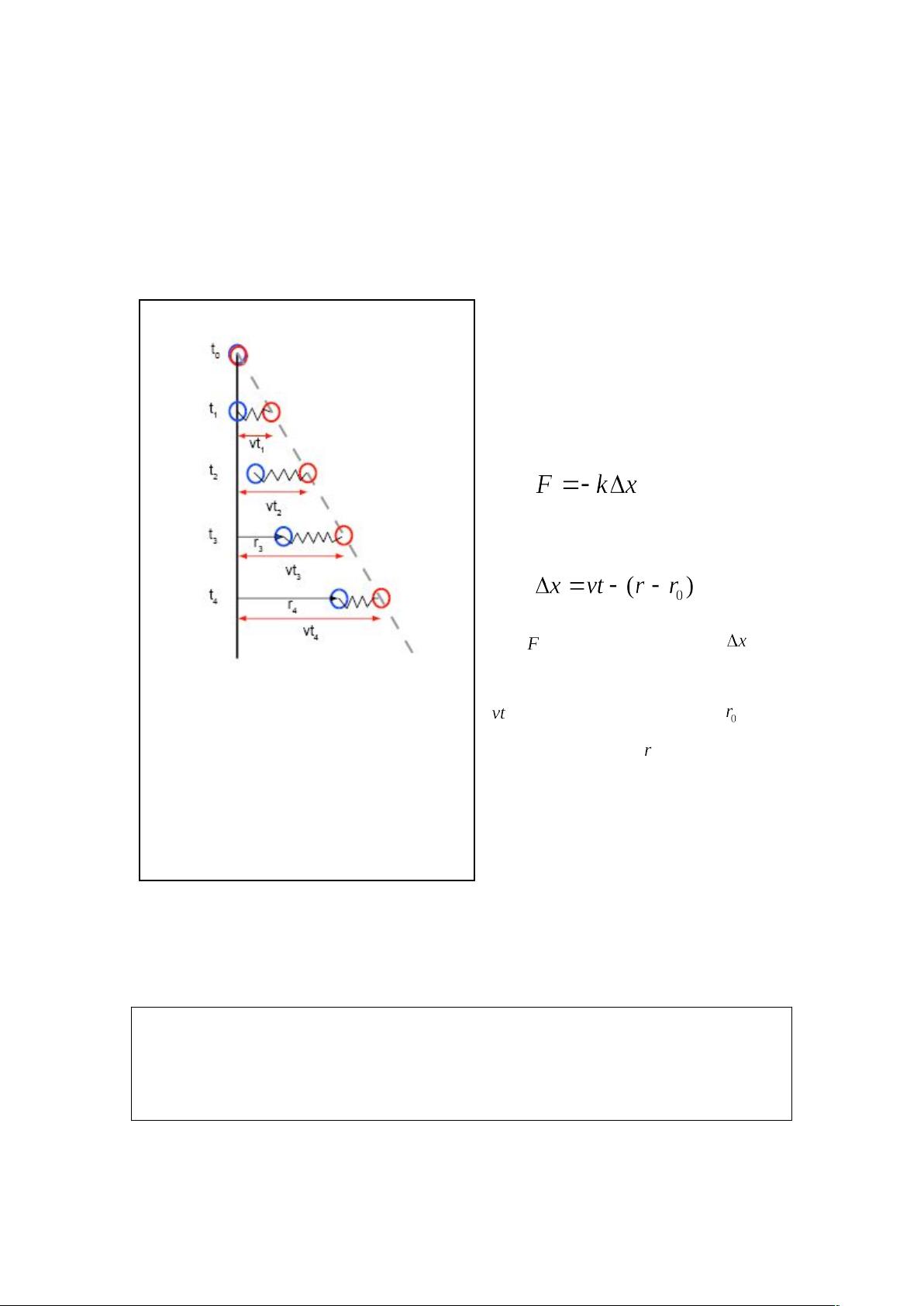
4 受控分子动力学模拟(Steered Molecular Dynamics)
所谓受控分子动力学模拟(Steered Molecular Dynamics,SMD),就是指在进行分
子动力学模拟时,人为地给分子中的某个或某几个原子施加一个假想的外力,或者人为地
固定某个或某几个原子的位置。从而控制整个分子的行为。我们前面进行的动力学模拟都
是通过各种参数设置,尽量逼近蛋白质分子在溶液体系中的真实状态,以研究分子的各种
性质和行为。但受控分子动力学模拟却要用一个假想的外力干扰控制生物大分子的行为,
这是为什么呢?(需要查一下更多的SMD的应用实例,详细了解SMD究竟可以用于研究什
么问题,才能做出回答)
在下面的例子中,我们将首先固定泛素分子中一个原子的位置,然后用一个假想的
外力牵拉另一个原子。球形的泛素分子会因此而被逐渐拉开,最终成为伸展状态的肽链。
进行SMD时,我们需要用已经完成能量最小化和能量平衡,达到稳定状态的蛋白结
构。如果用含有扭曲、拉伸、变形构象的原始蛋白结构,我们将无法分清蛋白各部分的运
动是由于蛋白内部的形变张力引起的,还是由于SMD实验附加的外力造成的。因此,进行
SMD之前必须预先进行一次平衡态分子动力学模拟,获得稳定结构。
这里,我们使用2.4节球状水体分子动力学模拟输出的恢复文件(.restart)进行SMD。
恢复文件输出时,泛素已在水体中完成了能量最小化和能量平衡,达到了稳定状态。
4.1 除去水分子
为了节省计算时间,我们在进行本次SMD之前将除去体系中所有的水分子。但读者
必须要注意:在真正进行SMD实验的时候决不可以将水分子除去!
1、打开VMD,选择File→New Molecule菜单项,载入common目录下的文件ubq_ws.
psf。不要关闭窗口,此时窗口“Load file for:”一项应该显示“0:ubq_ws.psf”。再次单击按钮
Browser,找到1-2-sphere目录下的文件ubq_ws_eq.restart.coor,载入该文件。关闭Molec
ule File Browser窗口。
现在我们载入了2.4节球状水体动力学模拟时输出的恢复文件(.restart.coor)。
2、选择Extension → tk菜单项,打开tk,首先用cd命令改变当前目录至common目录
下,然后输入:
这样我们便在文件夹下新建了一个文件,储存已经
达到平衡态的蛋白质分子,但没有水分子。



剩余26页未读,继续阅读
资源评论













