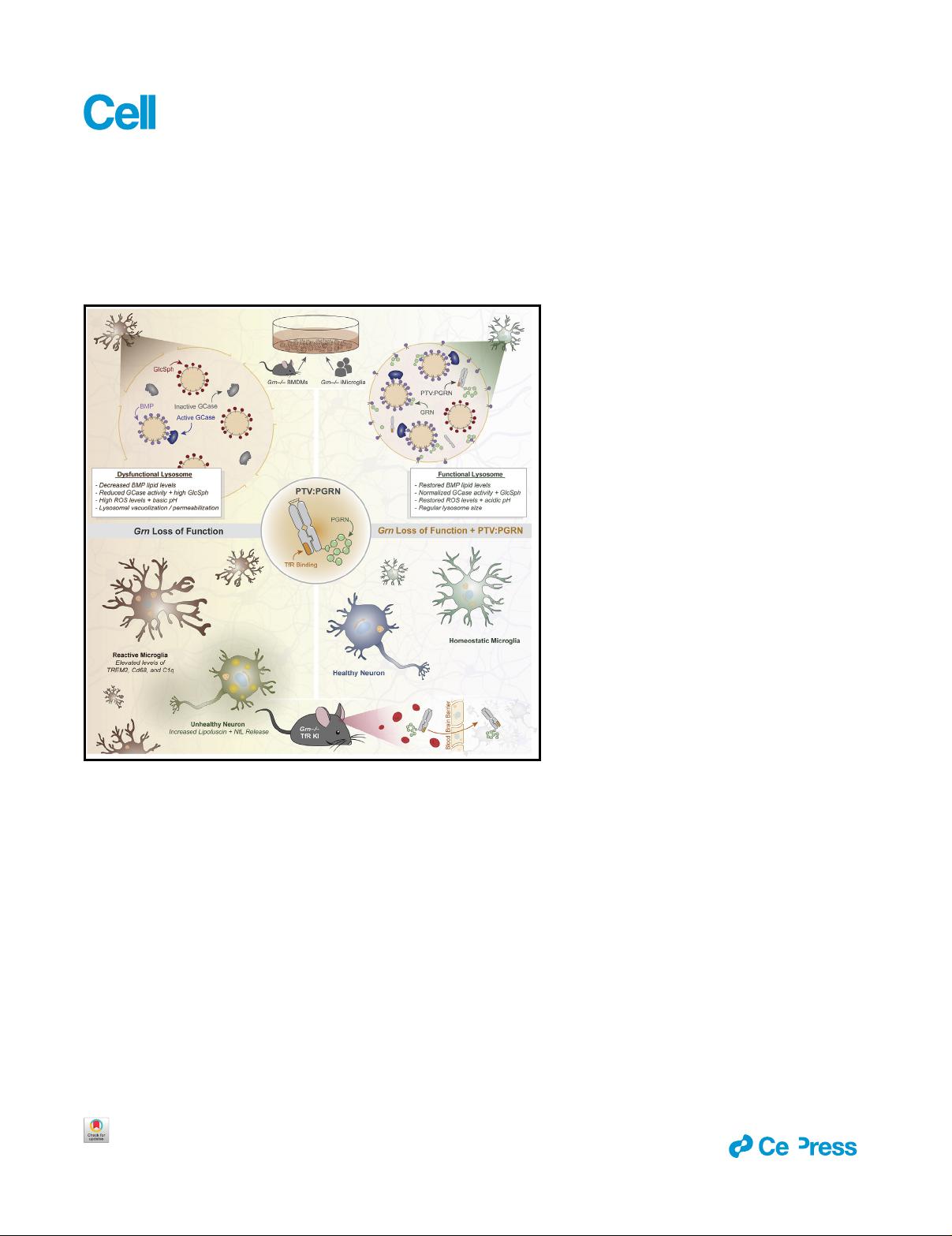
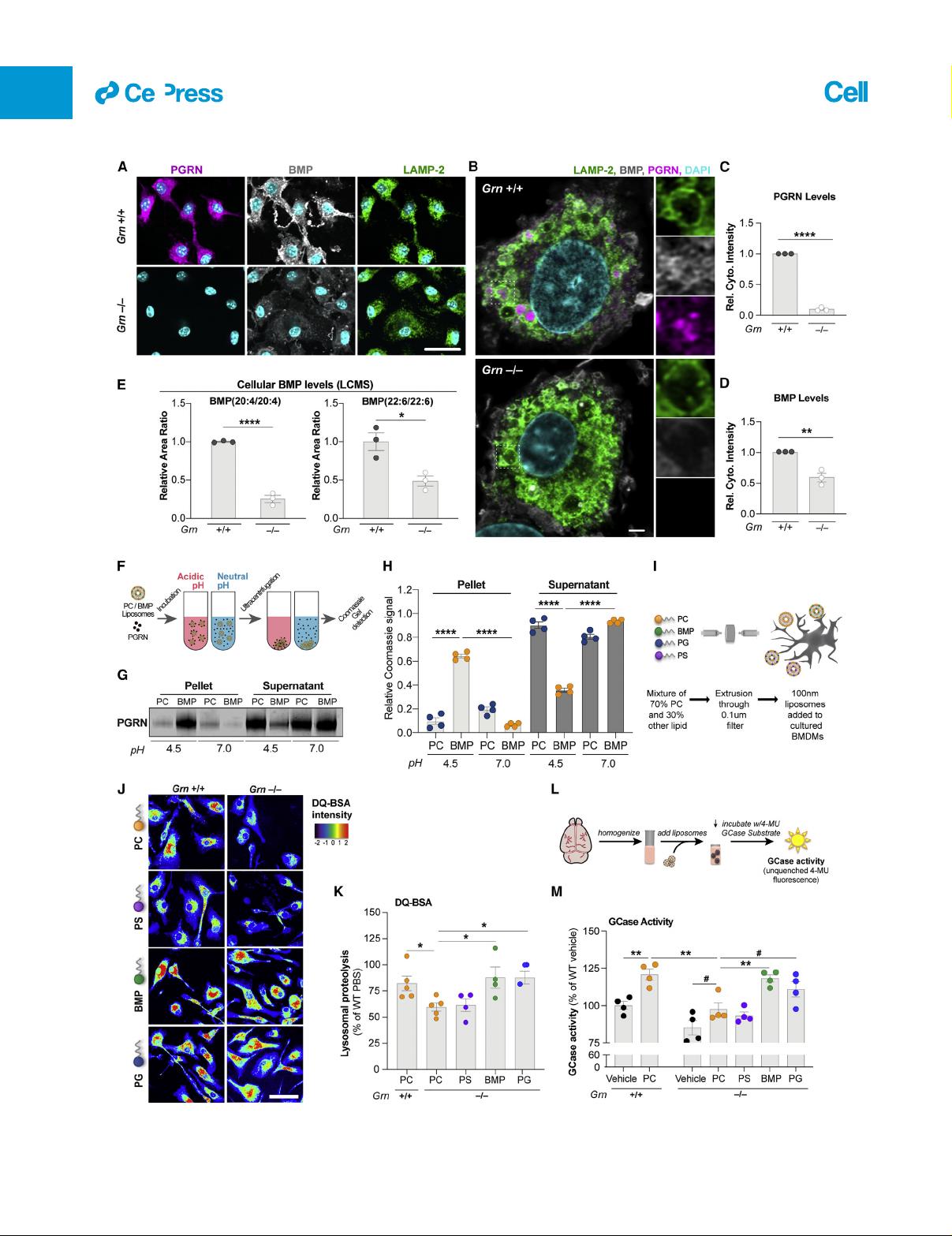
2019; Go
¨
tzl et al., 2014, 2018; Tanaka et al., 2017; Valdez et al.,
2017; Wils et al., 2012; Zhou et al., 2017a), lysosomal pH (Tanaka
et al., 2017), and glucocerebrosidase (GCase) activity, a lipase
involved in Gaucher disease and GBA-linked Parkinson disease
(Arrant et al., 2019; Jian et al., 2016; Valdez et al., 2020; Zhou
et al., 2019). PGRN was shown to regulate GCase activity via
direct binding (Jian et al., 2016), recruitment of co-chaperone
Hsp70 (Jian et al., 2016), and lysosomal delivery and processing
of prosaposin into saposin C, which activates GCase (Arra nt
et al., 2019; Paushter et al., 2018; Valdez et al., 2020; Zhou
et al., 2017c, 2019).
PGRN is processed intracellularly by CatB and CatL into seven
6 kDa granulin (GRN) peptides that may mediate its lysosomal
functions (Holler et al., 2017; Lee et al., 2017 ; Zhou et al., 2017b).
Given that key lysosomal PGRN interactors—CatD, prosaposin,
GCase—are genetically linked to neurodegeneration, defects in
lysosomal functions of PGRN or GRN peptides likely underlie
pathogenic mechanisms in GRN-FTD and NCL. This is corrobo-
rated by findings that a modifier of GRN-FTD age of onset is
TMEM106B (Cruchaga et al., 2011; Finch et al., 2011), a gene en-
coding a transmembrane lysosomal protein whose ablation
worsens lysosomal defects and FTD-like pathology of Grn
–/–
mice (Feng et al., 2020; Werner et al., 2020; Zhou et al., 2020).
In the CNS, Grn LoF profoundly impacts microglial state and
function (Go
¨
tzl et al., 2018, 2019; Kao et al., 2017; Krabbe
et al., 2017; Lui et al., 2016; Martens et al., 2012; Zhang et al.,
2020). Microglia express the highest CNS PGRN levels and
show higher expression in reactive states, as a result of injuries
(Byrnes et al., 2011; Moisse et al., 2009; Tanaka et al., 2013), ag-
ing (Zhang et al., 2020), or disease pathology (Chen-Plotkin et al.,
2010). Grn LoF in mice causes inflammation and microgliosis, a
hallmark of disease, along with enhanced neurotoxicity due in
part to secretion of complement factors (Go
¨
tzl et al., 2019;
Krabbe et al., 2017; Lui et al., 2016; Martens et al., 2012; Zhang
et al., 2020). Along with data showing Grn LoF microglia induce
aberrant TDP-43 in cultured neurons (Zhang et al., 2020), these
studies suggest microglial dysfunction may be pathogenic in pa-
tients. Since Grn
–/–
microglia share common features with other
LSD models, including expression profiles typical of reactive mi-
croglia (Colombo et al., 2021; Evers et al., 2017; Go
¨
tzl et al.,
2019; Ullman et al., 2020), lysosomal dysfunction may contribute
to driving the hyperreactive state of Grn LoF microglia. Because
lipofuscinosis is also a feature of PGRN-deficient neurons (Kao
et al., 2017; Paushter et al., 2018), we hypothesize that correct-
ing lysosomal dysfunction in both microglia and neurons in the
CNS of GRN-FTD patients may slow or arrest disease
progression.
Since GRN-FTD results from PGRN deficiency, a potentially
attractive therapeutic strategy is a CNS-penetrant PGRN
replacement therapy. Here, we capitalized on recently engi-
neered transport vehicles (TVs) that enable receptor-mediated
transcytosis of large molecules across endothelial cells of the
blood brain barrier (BBB) by binding to the human transferrin re-
ceptor apical domain (huTfR) (Kariolis et al., 2020; Ullman et al.,
2020). We fused a TV to recombinant human PGRN, termed
protein transport vehicle (PTV):PGRN, to assess efficacy in cor-
recting Grn LoF-associated phenotypes in vitro and in vivo. Our
results uncover a relationship between PGRN and lysosomal
lipid metabolism by defining the PGRN deficiency model as an
atypical LSD associated with a deficiency of the lysosomal lipid
bis(monoacylglycero)phosphate (BMP), leading to a secondary
storage of GCase substrates. Importantly, our data show
PTV:PGRN rescues a range of lysosomal defects, including
BMP deficiency, GCase substrate accumulation, lysosomal va-
cuolization and endomembrane damage, as well as inflamma-
tion and neuronal damage. Our data provide a pre-clinical proof
of concept supporting the use of PTV:PGRN for treatment of
GRN-FTD.
RESULTS
Grn
–/–
brains show lysosomal lipid alterations
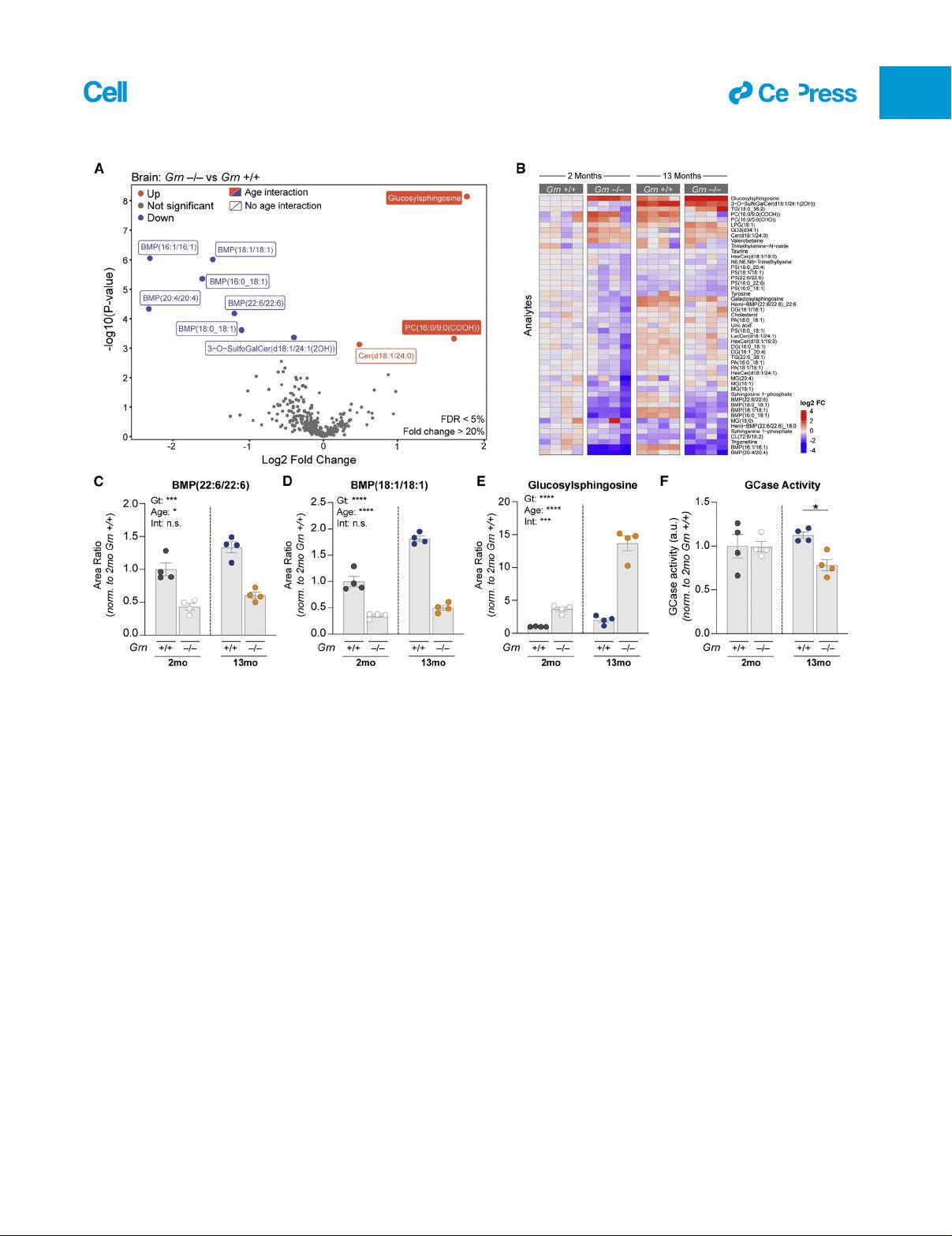
Previous liquid chromatography-mass spectrometry (LCMS)
studies have shown lipid metabolism is altered in various
Grn
–/–
models (Evers et al., 2017; Marschallinger et al., 2020).
However, lipid panels previously assessed were limited in
coverage of glycosphingolipids and other lipids often dysregu-
lated in LSDs, such as BMP (also known as lysobisphosphatidic
acid). To determine whether Grn LoF is associated with profiles
suggestive of LSDs, we performed targeted lipidomics and me-
tabolomics on young (2 month) and old (13 month) Grn
+/+
and
Grn
–/–
mouse brain with LCMS. We selected Grn
–/–
rather than
Grn
+/–
models, as they best replicate the GRN-FTD pathologies
(Kao et al., 2017). We found a decrease in various BMP species
including BMP(22:6/22:6) and BMP(18:1/18:1) as the largest ge-
notype-dependent changes (Figures 1A–1D). This anionic phos-
pholipid is enriched in late endosomes/lysosomes, where it is
believed to stimulate lysosomal lipases and cholesterol egress
into the cytosol (Breiden and Sandhoff, 2020; Gruenberg,
2020; McCauliff et al., 2019). BMP showed an age effect but
no age-genotype interaction (Figures 1C and 1D). Conversely,
there was a genotype- and age-dependent increase in brain
levels of glucosylsphingosine (GlcSph), a GCase substrate (Dai
et al., 2016; Taguchi et al., 2017)(Figures 1A, 1B, and 1E).
Consistent with GCase substrate accumulation, Grn
–/–
brain
showed a decrease in GCase activity, but only at 13 months ( Fig-
ure 1F). Grn
–/–
plasma, liver, and urine also revealed a decrease
in one or more BMP species, while Grn
–/–
plasma and liver ex-
hibited a GlcSph increase (Figures S1A–S1C). Overall, these
data establish BMP deficiency and GlcSph accumulation as
defining molecular features of Grn
–/–
mice and point to lysosomal
dysfunction.
Next, we analyzed plasma and cerebrospinal fluid (CSF) from
healthy controls and non-GRN FTD and GRN-FTD patients (Ta-
ble S1). PGRN deficiency was validated in the biofluids of GRN
mutation carriers (Figures S1 D and S1E). Neurofilament light
chain (Nf-L) increase was also confirmed in those diagnosed
with FTD (Figures S1F and S1G), indicative of neurodegeneration
(Ghidoni et al., 2008; Meeter et al., 2016). We found a subtle defi-
ciency in CSF BMP(18:1/18:1) (Figure S1H) and a decreasing
trend of BMP(22:6/22:6) in GRN-FTD CSF (Figure S1I) but not
in plasma (Figures S1J and S1K). Non-GRN FTD patients’ CSF
also exhibited a mild reduction of BMP(18:1/18:1) (Figure S1H),
suggesting potential converging lysosomal defects in FTD pa-
tients. GlcSph was significantly increased in GRN-FTD plasma
but not in non-GRN FTD plasma (Figure S1L), replicating the
ll
4652 Cell 184, 4651–4668, September 2, 2021
Article