class10气体在聚合体中扩散的测量实用PPT课件.pptx
版权申诉
102 浏览量
2021-10-06
22:29:47
上传
评论
收藏 1.97MB PPTX 举报

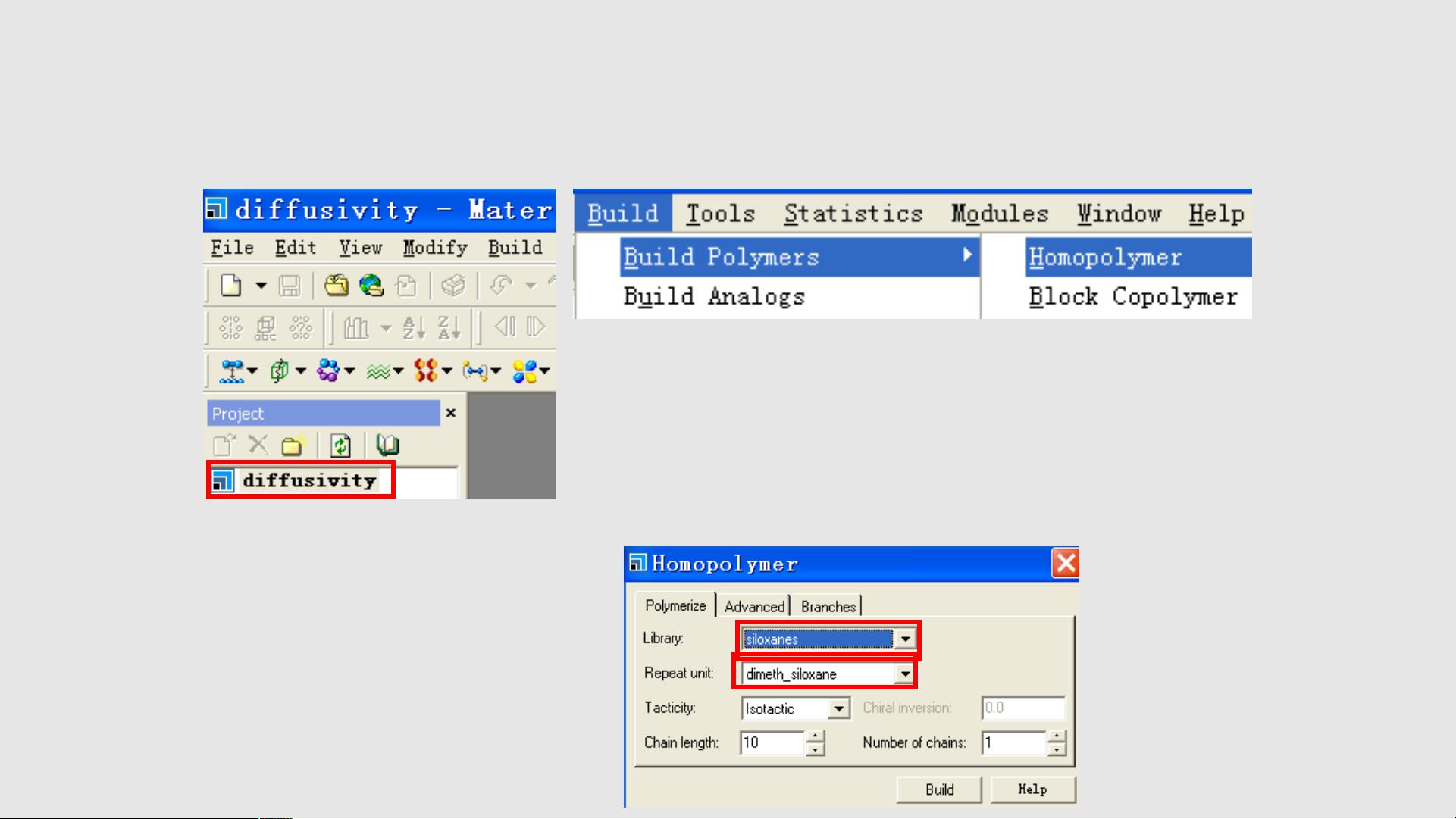
1. 建立初始结构
第一步是构建并优化氧分子和 PDMS 聚合物来构建无定形原胞。
从菜单栏中选择 Build / Build Polymers / Homopolymer 来显示
Homopolymer 对话框。
把库 Library 改成硅氧烷 siloxanes ,把重复单元 Repeat unit 改成二甲
基硅化物 dimeth_siloxane 。
第 1 页 / 共 39 页


剩余38页未读,继续阅读
资源评论


加油学习加油进步
- 粉丝: 1400
- 资源: 52万+

下载权益

C知道特权

VIP文章

课程特权
开通VIP











