论文笔记-Spatially informed cell-type deconvolution for spatial tran
需积分: 0 21 浏览量
2022-08-29
09:51:34
上传
评论
收藏 50.66MB DOCX 举报


论 文 笔 记 Spatially informed cell-type deconvolution for spatial
transcriptomics
(Nature Biotechnology 2022, 来自密西根大学)
论文摘要
许多空间分辨的转录组技术没有单细胞分辨率,而是从潜在的异质细胞类型的混合细胞中
测量每个点的平均基因表达。在这里,我们介绍了一种反褶积方法,即基于条件自回归的反褶
积(CARD),它将来自单细胞 RNA 测序(scRNA-seq)的细胞类型特异性表达信息与跨组织位置
的细胞类型组成的相关性结合起来。建模空间相关性可以使我们能够跨位置借用细胞类型的组
成信息,提高反褶积的准确性,即使有一个不匹配的 scRNA-seq 参考。CARD 还可以在未测
量的组织位置输入细胞类型组成和基因表达水平,从而构建一个精确的空间组织图,其分辨率
任意高于原始研究中的测量值,并可以在没有 scRNA-seq 参考的情况下进行反褶积。应用于
四个数据集,包括一个胰腺癌数据集,确定了多种细胞类型和具有不同空间定位的分子标记,
这些标记定义了胰腺癌的进展、异质性和区段化。
审阅人
占炎根
代码链接
www.xzlab.org/software.html
数据集
论文 code availability(很多)
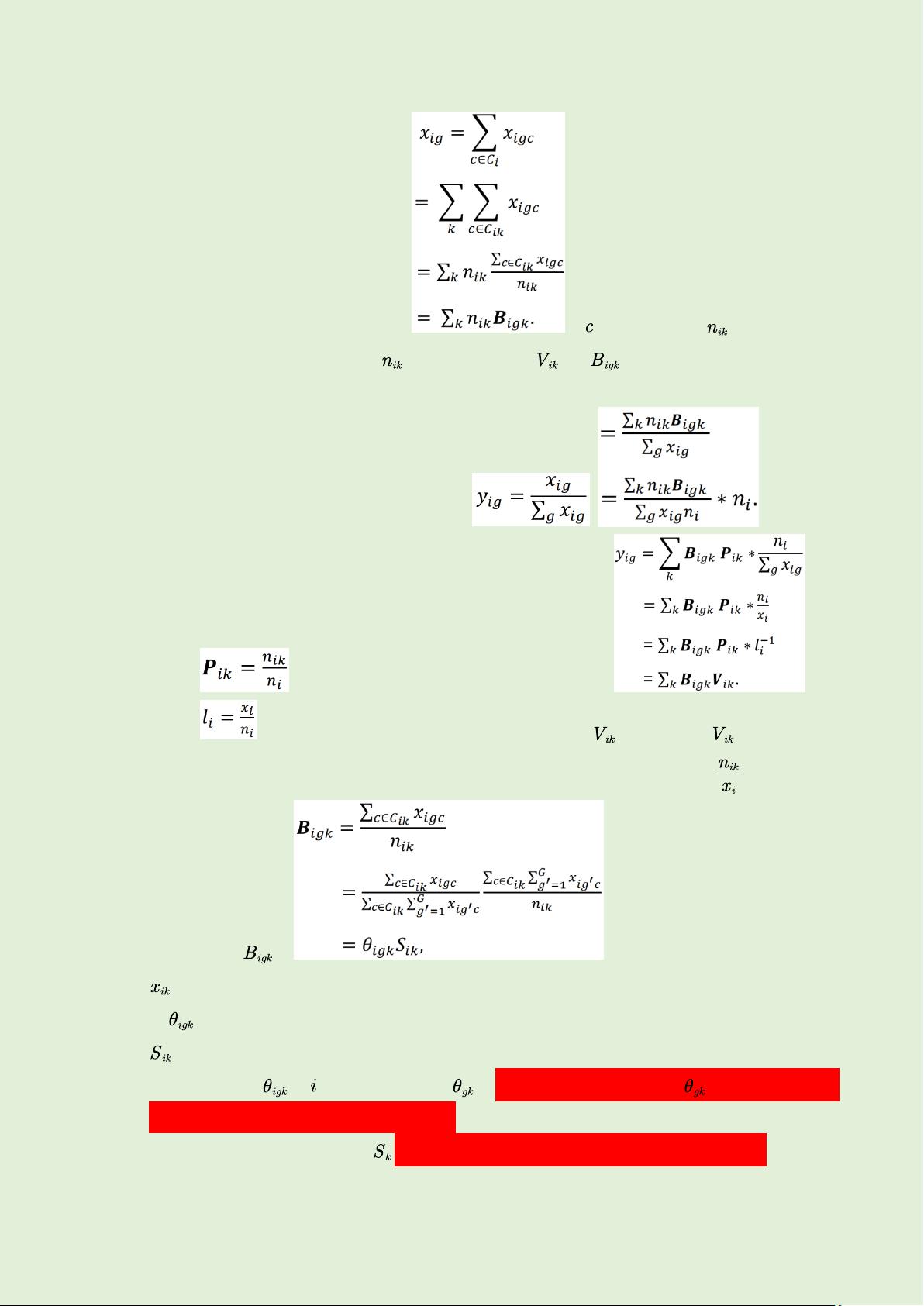
核心总览
its ability to accommodate the spatial correlation structure in cell-type
composition across tissue locations by a conditional autoregressive (CAR)
modeling assumption
enable accurate and robust deconvolution of spatial transcriptomics data across
technologies with different spatial resolutions and in the presence of
mismatched scRNA-seq references.
impute cell-type compositions as well as gene expression levels on new locations



剩余29页未读,继续阅读
资源评论













