





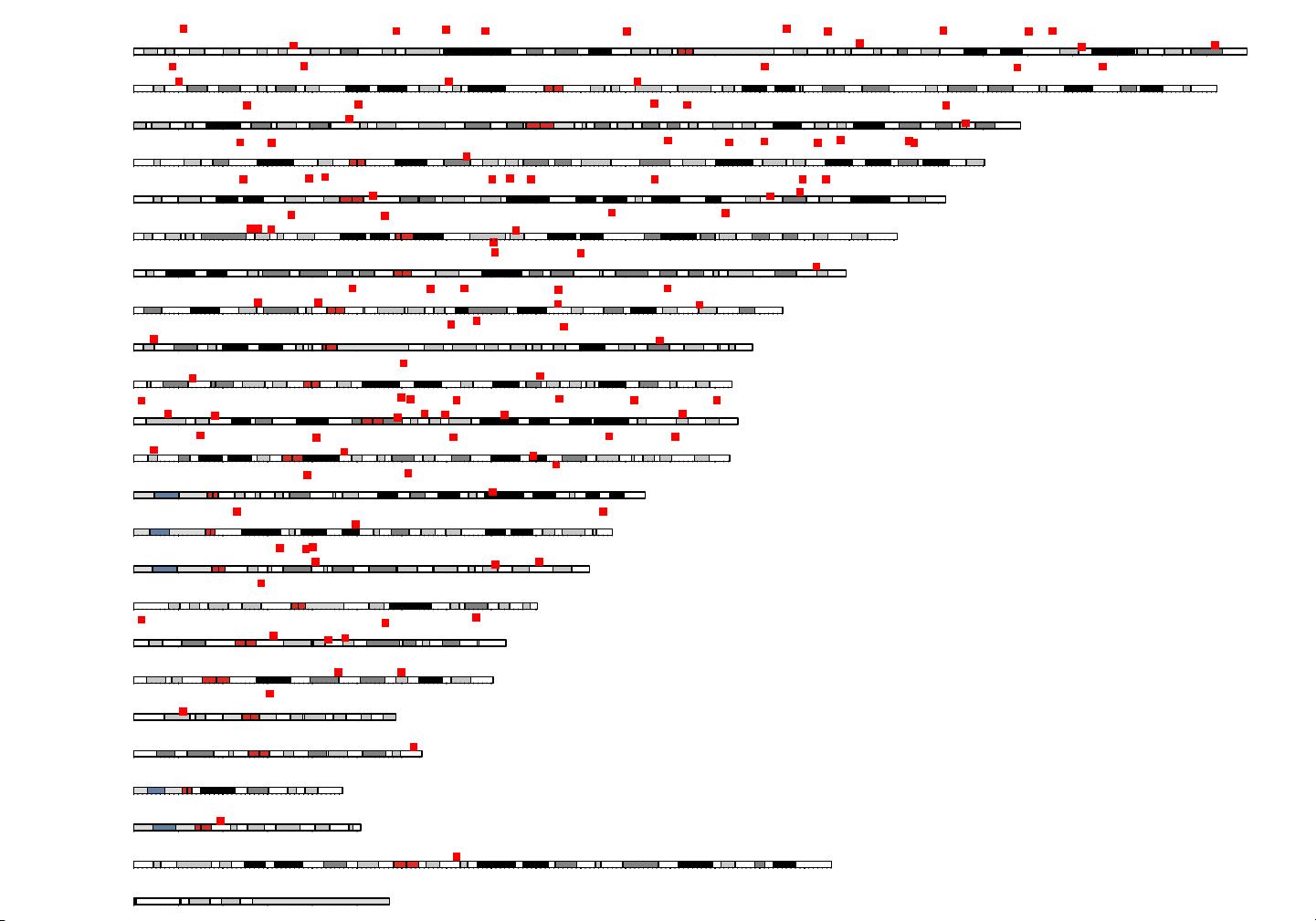
chr1
chr2
chr3
chr4
chr5
chr6
chr7
chr8
chr9
chr10
chr11
chr12
chr13
chr14
chr15
chr16
chr17
chr18
chr19
chr20
chr21
chr22
chrX
chrY
1 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 190 200 210 220 230 240
1 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 190 200 210 220 230 240
1 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 190
1 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 190
1 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180
1 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170
1 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150
1 10 20 30 40 50 60 70 80 90 100 110 120 130 140
1 10 20 30 40 50 60 70 80 90 100 110 120 130
1 10 20 30 40 50 60 70 80 90 100 110 120 130
1 10 20 30 40 50 60 70 80 90 100 110 120 130
1 10 20 30 40 50 60 70 80 90 100 110 120 130
1 10 20 30 40 50 60 70 80 90 100 110
1 10 20 30 40 50 60 70 80 90 100
1 10 20 30 40 50 60 70 80 90 100
1 10 20 30 40 50 60 70 80 90
1 10 20 30 40 50 60 70 80
1 10 20 30 40 50 60 70 80
1 10 20 30 40 50
1 10 20 30 40 50 60
1 10 20 30 40
1 10 20 30 40 50
1 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150
1 10 20 30 40 50














