全基因组关联研究(GWAS)已广泛应用于植物代谢组的复杂生物合成过程的研究。以往的研究大多采用单目遗传算法,如混合线性模型(MLM),对实现多位点遗传算法的更有效算法知之甚少。本文报道了6个多位点模型(FASTmrEMMA, FASTmrMLM, isisemi - blasso, mrMLM, pKWmEB, pLARmEB)
### 关键知识点解析
#### 全基因组关联研究(GWAS)及其实现方法
- **GWAS背景**: 全基因组关联研究是一种强大的工具,用于揭示特定性状或疾病与基因组变异之间的关联。这类研究对于理解复杂生物合成过程至关重要,尤其是在植物学领域。
- **传统方法限制**: 混合线性模型(Mixed Linear Model, MLM)是GWAS中常用的一种方法,但其基于Bonferroni校正的特性可能会忽略掉一些重要的遗传位点,因此寻找更为有效的多位点遗传算法成为当前研究的重点。
#### 多位点遗传算法介绍
文章提及了六种不同的多位点遗传算法,它们分别是:
1. **FASTmrEMMA** (Fast multi-locus random-SNP-effect EMMA): 这是一种快速的多位点随机SNP效应算法,特别适用于大型数据集。
2. **FASTmrMLM** (Fast multi-locus random-SNP-effect Mixed Linear Model): 该算法旨在提高MLM的效率,尤其在处理大规模数据时表现优异。
3. **isisemi-blasso** (Iterative Sure Independence Screening EM-Bayesian LASSO): 结合迭代确定独立筛选技术和贝叶斯LASSO算法,能够高效地筛选出重要变量。
4. **mrMLM** (multi-locus random-SNP-effect Mixed Linear Model): 这是一种基于MLM的多位点版本,能更好地识别与性状相关的多个位点。
5. **pKWmEB** (polygenic background control based Kruskal-Wallis test plus empirical Bayes): 结合非参数统计方法(Kruskal-Wallis检验)与经验贝叶斯方法,用于处理多基因背景下的复杂性状。
6. **pLARmEB** (polygenic background control based least angle regression plus empirical Bayes): 将最小角回归技术与经验贝叶斯方法结合,有效处理多基因背景。
#### 使用mrMLM进行GWAS分析
- **环境准备**:
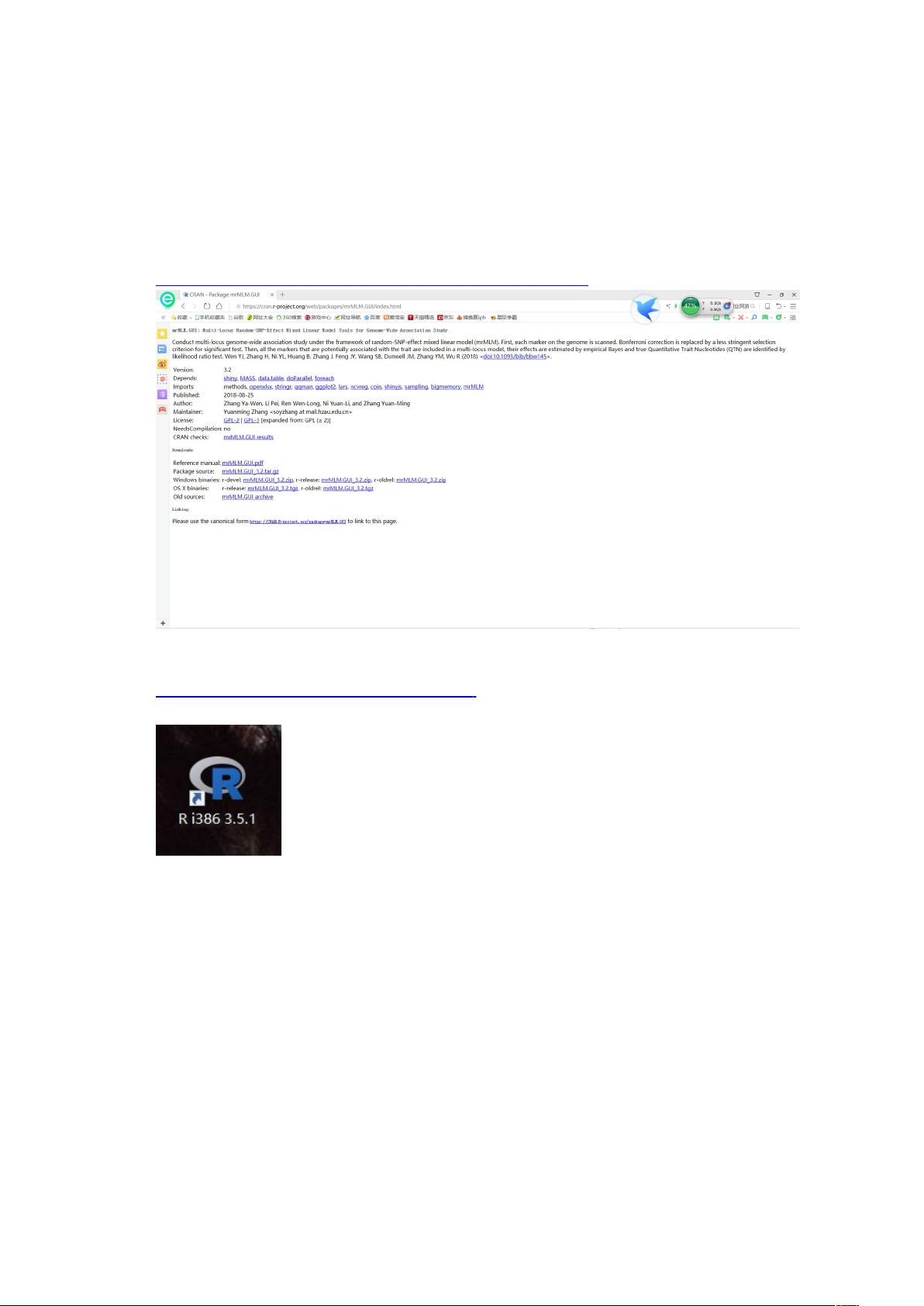
- 首先需要下载并安装R语言环境和mrMLM插件包。插件包可以从官方页面下载:`https://cran.r-project.org/web/packages/mrMLM.GUI/index.html`。
- R语言可以从清华镜像站点获取:`https://mirrors.tuna.tsinghua.edu.cn/CRAN/`。
- **加载mrMLM插件包**:
- 在R环境中安装并加载mrMLM插件包。这一步骤可以通过以下命令完成:`install.packages("mrMLM.GUI")` 和 `library(mrMLM)`。
- **运行mrMLM**:
- 使用mrMLM进行GWAS分析的基本语法如下:
```r
mrMLM(fileGen="路径", filePhe="路径", fileKin="路径", filePS="路径",
Genformat="格式", method=("方法"), Likelihood="方法", trait=范围,
SearchRadius=半径, CriLOD=阈值, SelectVariable=数量,
Bootstrap=TRUE, DrawPlot=TRUE, Plotformat="格式", Resolution="质量", dir="路径")
```
- 参数解释:
- `fileGen`: 基因型文件路径。支持.csv和.txt两种格式。文件中的数据可以是数字型、特征型或图谱型。
- `filePhe`: 表型文件路径。缺失值用NA表示。
- `fileKin`: 亲属关系矩阵文件路径。
- `filePS`: 种群结构文件路径。
- `Genformat`: 文件格式类型。“Num”代表数字型,“Cha”代表特征型,“Hmp”代表图谱型。
- `method`: 所选算法名称。例如:“mrMLM”、“FASTmrEMMA”等。
- `Likelihood`: 仅对FASTmrEMMA有效。可选项为“REML”或“ML”,分别代表限制最大似然估计和最大似然估计。
- `trait`: 要分析的性状编号范围。
- `SearchRadius`: 仅对mrMLM和FASTmrMLM有效,用于指定搜索半径(以kb为单位),即在多少kb范围内只选择一个可能关联的QTN。
- `CriLOD`: 显著QTN的关键LOD评分阈值。
- `SelectVariable`: 只适用于mrMLM,表示选择的变量数量。
- `Bootstrap`: 是否进行bootstrap验证。
- `DrawPlot`: 是否绘制结果图表。
- `Plotformat`: 图表格式。
- `Resolution`: 图表分辨率。
- `dir`: 结果输出目录路径。
通过上述步骤,用户可以利用mrMLM插件包有效地执行全基因组关联研究,并从中挖掘出与特定性状相关的遗传位点。这些方法不仅提高了GWAS的准确性,还极大地简化了复杂数据的处理流程,为生物学研究提供了强有力的支持。







评论1
最新资源