

Identification of microRNA precursor with the degenerate K-tuple
or Kmer strategy
Bin Liu
a,b,c,
n
, Longyun Fang
a
, Shanyi Wang
a
, Xiaolong Wang
a,b
, Hongtao Li
d
,
Kuo-Chen Chou
c,e
a
School of Computer Science and Technology, Harbin Institute of Technology Shenzhen Graduate School, Shenzhen, Guangdong, China
b
Key Laboratory of Network Oriented Intelligent Computation, Harbin Institute of Technology Shenzhen Graduate School, Shenzhen, Guangdong, China
c
Gordon Life Science Institute, Boston, MA 0478, USA
d
Wendeng Marine Environmental Monitoring Station, Station Oceanic Administration Wendeng, Weihai, Shandong, China
e
Center of Excellence in Genomic Medicine Research (CEGMR), King Abdulaziz University, Jeddah 21589, Saudi Arabia
HIGHLIGHTS
microRNA (miRNA) plays an important role in gene expression.
Identification of real pre-miRNAs is important miRNA-based therapy.
A novel predictor was developed for fast and effectively identifying miRNA.
article info
Article history:
Received 16 July 2015
Received in revised form
21 August 2015
Accepted 24 August 2015
Available online 9 September 2015
Keywords:
MicroRNA precursor
True pre-miRNA
False pre-miRNA
Degenerate Kmer
deKmer web-server
Long-range effect
abstract
The microRNA (miRNA), a small non-coding RNA molecule, plays an important role in transcriptional and
post-transcriptional regulation of gene expression. Its abnormal expression, however, has been observed
in many cancers and other disease states, implying that the miRNA molecules are also deeply involved in
these diseases, particularly in carcinogenesis. Therefore, it is important for both basic research and
miRNA-based therapy to discriminate the real pre-miRNAs from the false ones (such as hairpin
sequences with similar stem-loops). Most existing methods in this regard were based on the strategy in
which RNA samples were formulated by a vector formed by their Kmer components. But the length of
Kmers must be very short; otherwise, the vector's dimension would be extremely large, leading to the
“high-dimension disaster” or overfitting problem. Inspired by the concept of “degenerate energy levels”
in quantum mechanics, we introduced the “degenerate Kmer” (deKmer) to represent RNA samples. By
doing so, not only we can accommodate long-range coupling effects but also we can avoid the high-
dimension problem. Rigorous jackknife tests and cross-species experiments indicated that our approach
is very promising. It has not escaped our notice that the deKmer approach can also be applied to many
other areas of computational biology. A user-friendly web-server for the new predictor has been
established at http://bioinformatics.hitsz.edu.cn/miRNA-deKmer/, by which users can easily get their
desired results.
& 2015 Elsevier Ltd. All rights reserved.
1. Introduction
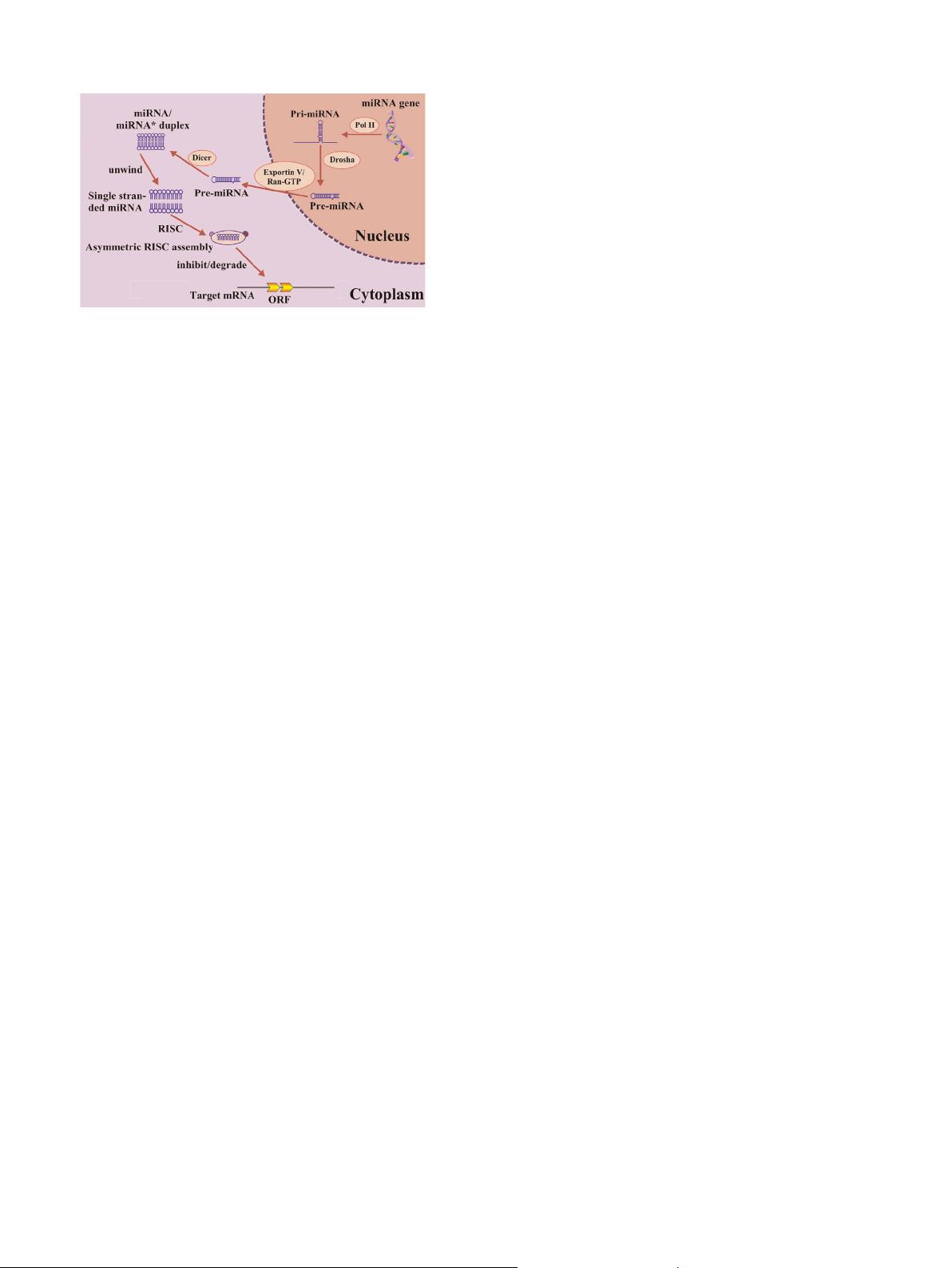
MicroRNAs (miRNAs) are small single-strand and non-coding
RNAs (ncRNAs), which play important roles in gene regulation by
targeting messenger RNAs (mRNAs) for cleavage or translational
repression (Fig.1). Their lengths are about 17–25 nt (Lopes et al.,
2014). The miRNAs are also involved in many important biological
processes, such as affecting stability, translation of mRNAs, and
negatively regulating gene expression in post-transcriptional
processes. Therefore, it is fundamentally important to identify the
real pre-miRNAs from the false ones. Unfortunately, it is difficult to
Contents lists available at ScienceDirect
journal homepage: www.elsevier.com/locate/yjtbi
Journal of Theoretical Biology
http://dx.doi.org/10.1016/j.jtbi.2015.08.025
0022-5193/& 2015 Elsevier Ltd. All rights reserved.
n
Correspondence to: Harbin Institute of Technology Shenzhen Graduate School,
HIT Campus Shenzhen University Town, Xili, Shenzhen 518055, China.
Tel.: þ 86 0755 2603 3283.
E-mail addresses: bliu@insun.hit.edu.cn (B. Liu),
dragoncloudest@gmail.com (L. Fang), wangshanyiwsy@gmail.com (S. Wang),
wangxl@insun.hit.edu.cn (X. Wang), lht760@126.com (H. Li),
kcchou@gordonlifescience.org (K.-C. Chou).
Journal of Theoretical Biology 385 (2015) 153–159
剩余6页未读,继续阅读
资源评论


weixin_38563525
- 粉丝: 4
- 资源: 966









最新资源
- 打火机识别检测数据集yolov11格式.zip
- 基于JAVA+SpringBoot+Vue+MySQL的在线英语阅读分级平台源码+数据库+论文(高分毕业设计).zip
- 基于JAVA+SpringBoot+Vue+MySQL的疫情物资捐赠和分配系统源码+数据库+论文(高分毕业设计).zip
- 基于JAVA+SpringBoot+Vue+MySQL的医院信息管理系统源码+数据库(高分毕业设计).zip
- 微信PC端的个人免签收款监控和支付回调,永不掉线,稳定易语言开源开源.zip
- 包含PyQt窗口布局常用控件程序
- 基于JAVA+SpringBoot+Vue+MySQL的智慧养老中心管理系统源码+数据库(高分毕业设计).zip
- 基于JAVA+SpringBoot+Vue+MySQL的自习室管理和预约系统源码+数据库+论文(高分毕业设计).zip
- 基于JAVA+SpringBoot+Vue+MySQL的智能停车计费系统源码+数据库(高分毕业设计).zip
- 无11111111111111111111111标题
- 微信小程官网,前后端源码都提供,并提供完善小程序学习资料 基于小程序wepy语言,具备cms网站的基本功能,能够打造简单易用的公司官网.zip
- Android arm64 hexdump tool ,源码,Android.mk 及编译好的bin
- 深入解析YOLO系列目标检测:头部(Head)结构与输出内容
- BDCI 2024基于TPU平台的OCR模型性能优化源码.zip
- 我写的各种易语言代码都会发到这里.zip
- 清华大学小学期LLM课程笔记:基础部分
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


