实验工作和基因组序列分析揭示了在细胞中RNA有广泛的作用[15]。除了遗传信息的传输,
非编码RNA能催化生物化学反应,并在多种调节过程中有关键作用。还有一些功能RNA在它
们的单链信息或是RNA蛋白质复合物环境下表现很重要,另一方面,功能是与RNA的三维
结构直接相关[6]。获取这些结构知识对理解其功能非常重要。然而,通过实验测定RNA的结
构仍然具有挑战性。在RCSB的蛋白质数据库(ProteinDataBank,PDB)[7]里的全部结构
数据中,只有少于6%的数据包含RNA。由于测序技术的进步,许多在不同生物体里RNA已
经被测序,并分类到同源家族的Rfam数据库中[8]。然而,在目前列出的2450个家族里,只
有59(2.4%)个包含至少一个PDB结构数据,然而却有954(39%)个包括超过100条序
列,甚至566(23%)个超过200条序列。因此为了充分使用海量的序列信息,计算机辅助
结构预测方法是一个很有前景的互补技术。不幸的是,目前最先进的计算机辅助方法在未知
RNA结构预测上也很难使RMSD(均方根差)值降到812埃[910],远高于能达到23埃精度
的X射线晶体结构。通常情况下,计算机辅助方法[1113]局限于众多限制,包括结果不定,
或是要求专家参与,复杂的拓扑结构不能够可靠预测[1415]。一些在此方向上获得的最好的
结果是通过结合计算机辅助和实验方法:突变研究得到的跨残基关联作为约束加入到计算模
型[1617]。
在相关的蛋白质结构预测领域,近年通过探究残基协同进化,在残基关联预测方面有了显著
的进步[18]。这些新方法基于两种思路:(i)两残基(甚至可以在一级序列上相距很远)间的
三级结构关联导致氨基酸占位的相关模式,这个可以被多序列比对(MultipleSequence
Alignments,MSA)统计分析方法检测到。(ii)本地相关性测量,比如相互作用信息
(MI),因为传递效应而令人困惑:即使不直接连接而是通过相邻联系到一个共同的中间残
基的两个位置也将相关。像DCA[19,20]和与之类似的方法[21,22]它们的优点是能够区分直接
和间接效果来推断间接相关的直接耦合作用。这导致在预测关联数据的精度大幅提高,可用
于预测三级和四级蛋白质结构,例如球蛋白[23,24],蛋白质复合物[2527],积极构象[28]或
膜蛋白[29]。
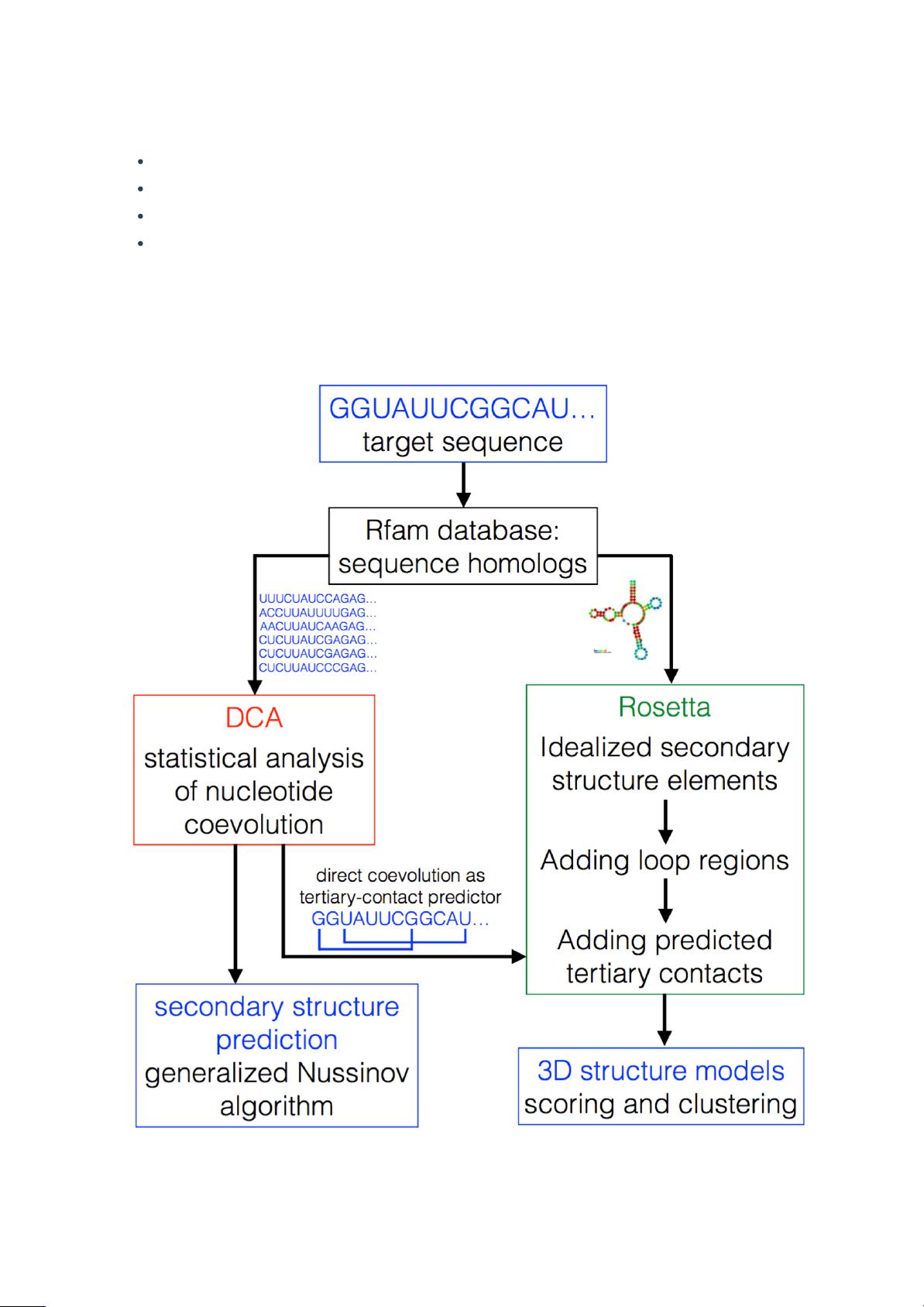
我们的目标是提出一个有效的基于协同进化分析现有的同源序列的RNA二级结构和三级结构
预测流程。用协方差模型进行比较RNA序列分析是一致公认的[30,31]:MI能够成功的用来推
断基本配对和预测二级结构[30,32,33]。尽管已经观察的一些三级结构间的联系表明MI的重
要性[32,33],然而,有人认为,在三级结构中存在关联的非规范碱基对较少表现出协变
性[35]。这里我们发现,在将本地协方差分析方法换为DCA时,这些微弱的信号却能在三维
结构关联上显著增强。使用经过严格筛选的具有复杂结构的核糖开关家族数据集,我们发现
DCA能够有效的整合进现有的RNA二级结构和三级结构预测的工具中。我们表明,结合类似
于Nussinov算法之类的标准方法[36],相比基于MI的预测方法DCA能系统的提高二级结构的
预测程度。将DCA整合到Rosetta中[37],一个非常成功的RNA结构预测工具[10],我们提出
了一个完全自动化的三级结构预测方案。相对于仅仅使用Rosetta,其结果有显著提高,并
且比那些RNApuzzle实验中包含的工作流更具有竞争力,因为那些方法部分的需要额外的实
验信息或者是专家的操作[9,10]。
材料与方法
推论流程