

实验四 基于 Matlab 的序列比对分析
实验目的
1. 了解MATLAB7.x生物信息工具箱中的序列比对方法;
2.熟悉从数据库获取序列信息, 查找序列的开放阅读框, 将核普酸序列转
换为氨基酸序列, 绘制比较两氨基酸序列的散点图, 用Needleman-wunsch算法和
Smith-Waterman算法进行比对, 以及计算两序列的同一性的方法;
3.熟悉与序列比对相关的生物信息学函数。
所需软件
MATLAB 7.0或 MATLAB 7.0 以上的版本
实验内容
序列比对是生物信息学的重要基础。进行序列比对的目的之一是判断两个序
列之间是否具有足够的相似性,从而判定二者之间是否具有同源性。序列比对的
基本算法主要有两个,一个是用于全局比对的 Needleman-Wunsch 算法,另一个
是主要用于局部比对的 Smith-Waterman 算法,而后者又是在前者的基础上发展
起来的。在 MATLAB生物信息工具箱中,序列比对主要用这两种算法。
确定两个序列的相似性是生物信息学的基础工作,通过序列比对(又称序列
联配),可以确定两个序列是否具有同源性。
1. 查找序列信息
Tay-Sachs症是一种由于缺乏 ß-氨基已糖苷酶 A(Hex A)而导致的常染色体
隐性遗传疾病。这种酶能分解大脑和神经细胞中的神经节苷脂(GM2)。基
因 HEXA 编码该酶的 ß 亚基,而第三个基因 GM2A 编码活化剂蛋白质 GM2。
1.1 查找目的基因 Tay-Sachs
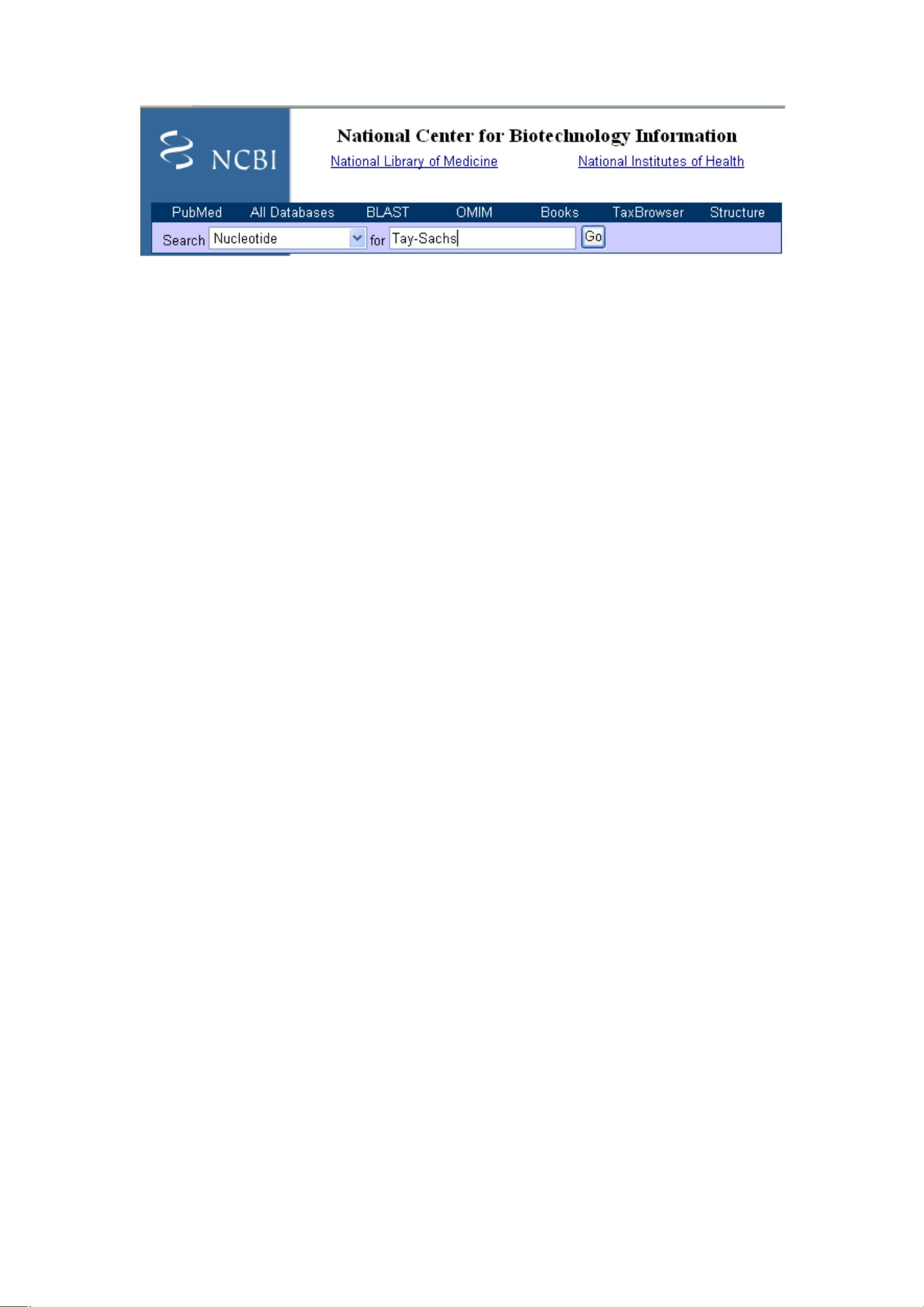
在 NCBI(http://www.ncbi.nlm.nih.gov)上查找信息,在 Search 列表中选择
[Nucleotide],在 for 框中输入[Tay-Sachs], 点击 Go。

剩余10页未读,继续阅读
资源评论


苦茶子12138
- 粉丝: 1w+
- 资源: 7万+

下载权益

C知道特权

VIP文章

课程特权
开通VIP









最新资源
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


