

Materials Studio 案例 1:
Au(111)表面自组装单层膜结构优化
目的:用 Materials Studio(MS)软件对金表面自组装膜的结构进行优化。
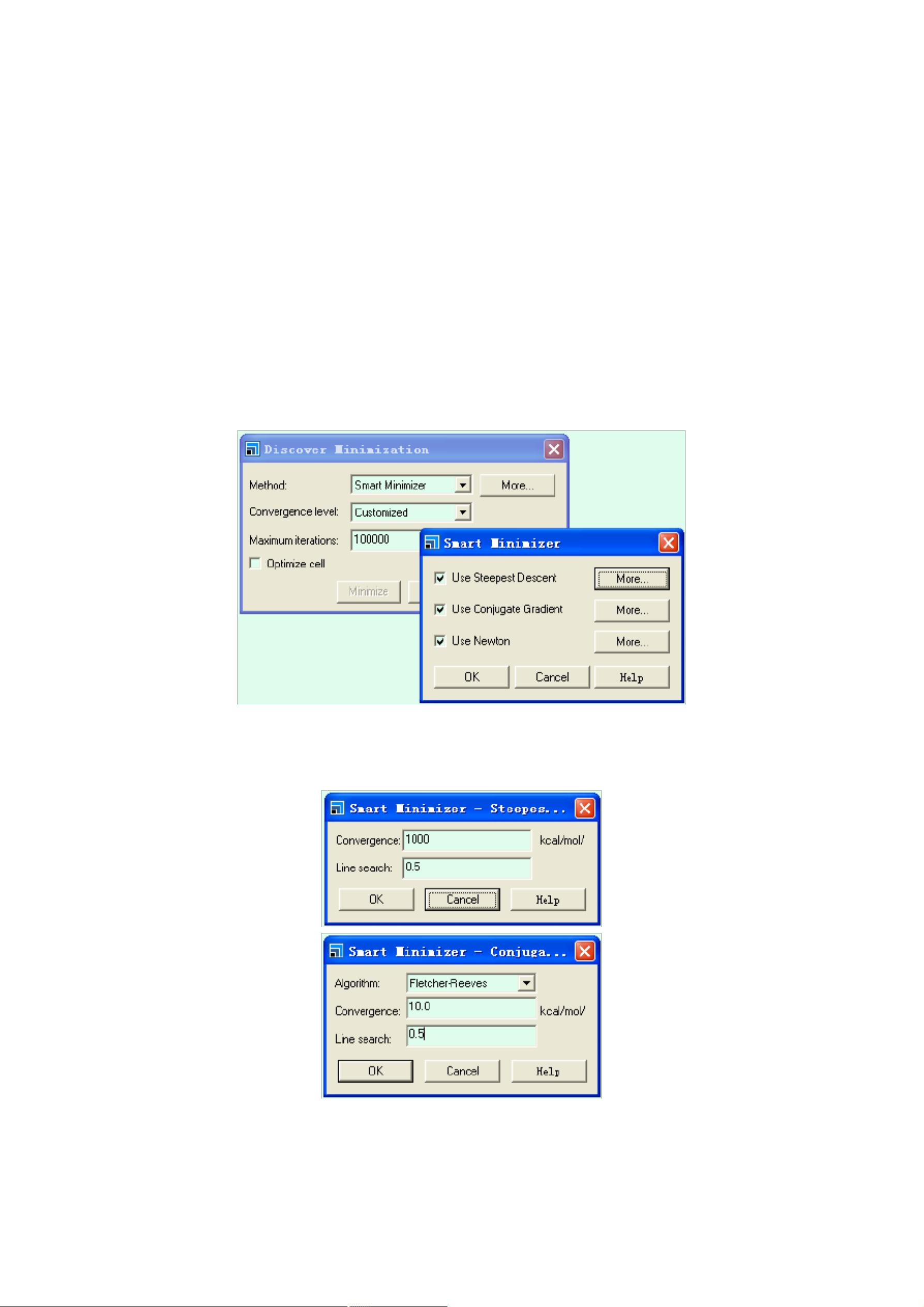
模块:Minimizer
MS Discover 结构优化原理
分子的势能一般为键合(键长、键角、二面角、扭转角等)和非键合相互作
用(静电作用、范德华作用等)能量项的加和,总势能是各类势能之和,如下式:
总势能 = 范德华非键结势能 + 键伸缩势能 + 键角弯曲势能
+ 双面角扭曲势能 + 离平面振动势能 + 库伦静电势能 + …
除了一些简单的分子以外,大多数的势能是分子中一些复杂形势的势能的组
合。势能为分子中原子坐标的函数,由原子不同的坐标所得到的势能构成势能面
(Potential Energy Surface,PES)。势能越低,构象越稳定,在系统中出现的
机率越大;反之,势能越高,构象越不稳定,在系统中出现的机率越小。通常势
能面可得到许多极小值的位置,其中对应于最低能量的点称为全局最小值
(Global Energy Minimum),相当于分子最稳定的构象。由势能面求最低极小值
的过程称为能量最小化( Energy Minimum ),其所对应的结构为最优化结构
(Optimized Structure),能量最小化过程,亦是结构优化的过程。
通过最小化算法进行结构优化时,应避免陷入局部最小值(local minimum),
也就是避免仅得到某一构象附近的相对稳定的构象,而力求得到全局最小值,即
实现全局优化。分子力学的最小化算法能较快进行能量优化,但它的局限性在于
易陷入局部势阱,求得的往往是局部最小值,而要寻求全局最小值只能采用系统
搜寻法或分子动力学法。在 Materials Studio 的 Discover 模块中,能量最小化
算法有以下四种:
1)最陡下降法(Steepest Descent),为一经典的方法,通过迭代求导,对
多变量的非线性目标函数极小化,按能量梯度相反的方向对坐标添加一位移,即
能量函数的负梯度方向是目标函数最陡下降的方向,所以称为最陡下降法。此法
计算简单,速度快,但在极小值附近收敛性不够好,造成移动方向正交。最陡下

剩余11页未读,继续阅读
资源评论

 2301_767299142023-03-07果断支持这个资源,资源解决了当前遇到的问题,给了新的灵感,感谢分享~
2301_767299142023-03-07果断支持这个资源,资源解决了当前遇到的问题,给了新的灵感,感谢分享~











