
B
B
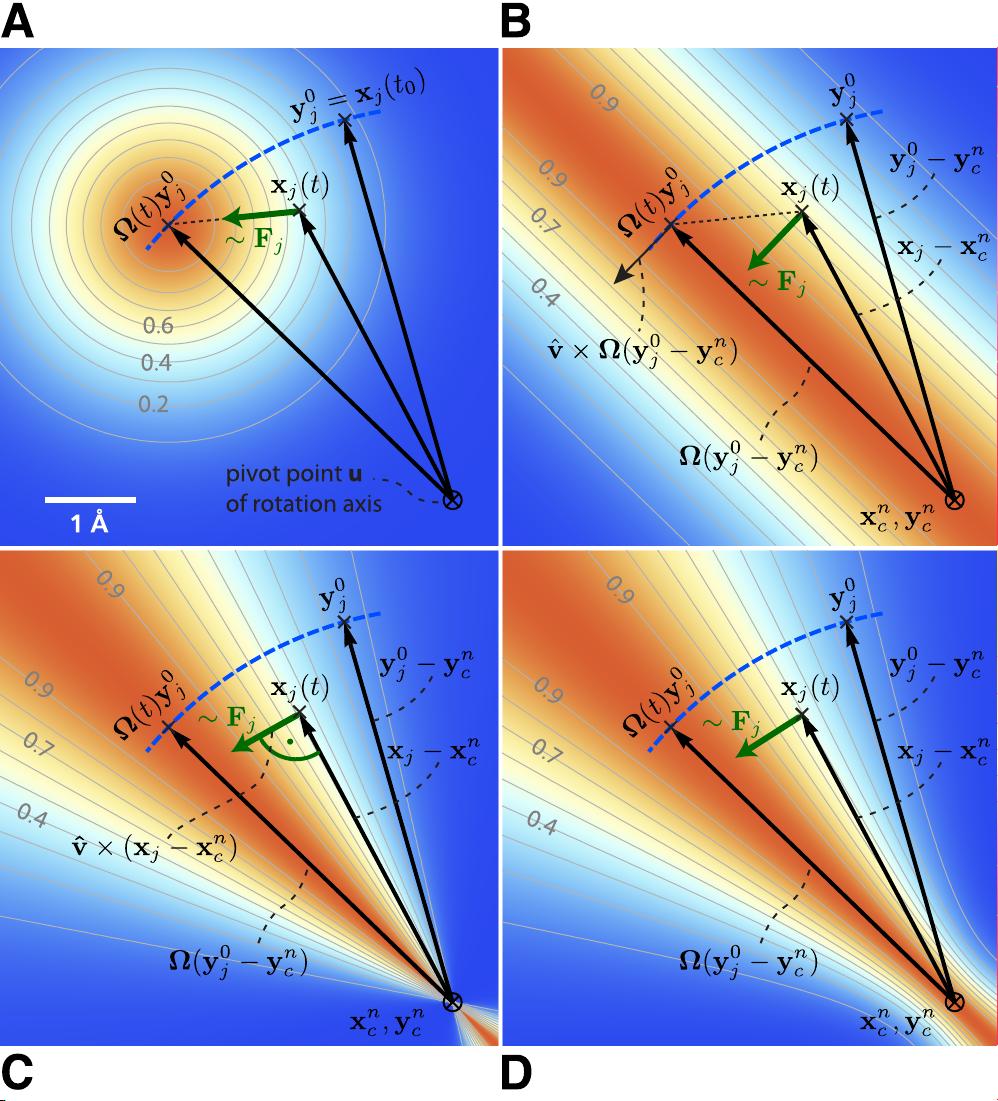
A B C
U [V]
z
A
0 0.4 0.8
2 nm
q
ref
4 e
8 e
12 e
0 e
U
channel 1
channel 0
 gromacs-5.0.7 _linux.tar.gz (2000个子文件)
gromacs-5.0.7 _linux.tar.gz (2000个子文件)  tng_io.c 648KB domdec.c 302KB nbnxn_search.c 187KB genborn_allvsall_sse2_single.c 181KB nb_kernel_ElecCSTab_VdwCSTab_GeomW4W4_avx_256_single.c 173KB pme.c 170KB nb_kernel_ElecEwSw_VdwLJSw_GeomW4W4_avx_256_double.c 168KB readir.c 164KB nb_kernel_ElecCSTab_VdwCSTab_GeomW3W3_avx_256_single.c 163KB bondfree.c 160KB nb_kernel_ElecEwSw_VdwLJSw_GeomW4W4_sse2_single.c 160KB nb_kernel_ElecEwSw_VdwLJSw_GeomW4W4_sse4_1_single.c 159KB nb_kernel_ElecEwSw_VdwLJSw_GeomW3W3_avx_256_double.c 158KB nb_kernel_ElecCSTab_VdwLJ_GeomW4W4_avx_256_single.c 157KB gmx_hbond.c 154KB nb_kernel_ElecEwSw_VdwNone_GeomW4W4_avx_256_double.c 154KB nb_kernel_ElecEwSw_VdwLJSw_GeomW4W4_sse4_1_double.c 154KB nb_kernel_ElecEwSw_VdwNone_GeomW3W3_avx_256_double.c 154KB nb_kernel_ElecEwSw_VdwLJSw_GeomW4W4_avx_256_single.c 154KB nb_kernel_ElecEwSw_VdwLJSw_GeomW4W4_sse2_double.c 154KB nb_kernel_ElecEwSw_VdwLJSw_GeomW4W4_sparc64_hpc_ace_double.c 152KB nb_kernel_ElecCSTab_VdwLJ_GeomW3W3_avx_256_single.c 150KB nb_kernel_ElecEwSw_VdwLJSw_GeomW3W3_sse4_1_single.c 150KB nb_kernel_ElecEwSw_VdwLJSw_GeomW3W3_sse2_single.c 150KB nb_kernel_ElecEwSw_VdwLJSw_GeomW4W4_avx_128_fma_double.c 150KB nb_kernel_ElecCSTab_VdwNone_GeomW4W4_avx_256_single.c 147KB nb_kernel_ElecCSTab_VdwNone_GeomW3W3_avx_256_single.c 147KB nb_kernel_ElecEwSw_VdwNone_GeomW4W4_sse4_1_single.c 146KB nb_kernel_ElecEwSw_VdwNone_GeomW4W4_sse2_single.c 146KB nb_kernel_ElecEwSw_VdwNone_GeomW3W3_sse4_1_single.c 146KB nb_kernel_ElecEwSw_VdwNone_GeomW3W3_sse2_single.c 146KB nb_kernel_ElecEwSw_VdwLJSw_GeomW3W3_sse4_1_double.c 145KB nb_kernel_ElecEwSw_VdwLJSw_GeomW3W3_sse2_double.c 144KB nb_kernel_ElecEwSw_VdwLJSw_GeomW3W3_avx_256_single.c 143KB nb_kernel_ElecEwSw_VdwLJSw_GeomW3W3_sparc64_hpc_ace_double.c 143KB pull_rotation.c 142KB nb_kernel_ElecEwSw_VdwLJSw_GeomW3W3_avx_128_fma_double.c 141KB nb_kernel_ElecEwSw_VdwNone_GeomW4W4_sse4_1_double.c 141KB nb_kernel_ElecEwSw_VdwNone_GeomW3W3_sse4_1_double.c 141KB nb_kernel_ElecEwSw_VdwNone_GeomW4W4_sse2_double.c 140KB nb_kernel_ElecEwSw_VdwNone_GeomW3W3_sse2_double.c 140KB nb_kernel_ElecEwSw_VdwNone_GeomW4W4_avx_256_single.c 140KB nb_kernel_ElecEwSw_VdwNone_GeomW3W3_avx_256_single.c 139KB nb_kernel_ElecEwSw_VdwNone_GeomW4W4_sparc64_hpc_ace_double.c 139KB nb_kernel_ElecEwSw_VdwNone_GeomW3W3_sparc64_hpc_ace_double.c 139KB nb_kernel_ElecEwSw_VdwNone_GeomW4W4_avx_128_fma_double.c 137KB nb_kernel_ElecEwSw_VdwNone_GeomW3W3_avx_128_fma_double.c 137KB nb_kernel_ElecEwSh_VdwLJEwSh_GeomW4W4_avx_256_double.c 137KB nb_kernel_ElecCSTab_VdwCSTab_GeomW4W4_avx_256_double.c 134KB gmx_arpack.c 133KB nb_kernel_ElecEwSh_VdwLJSh_GeomW4W4_avx_256_double.c 131KB nb_kernel_ElecEwSw_VdwLJSw_GeomW4W4_avx_128_fma_single.c 130KB nb_kernel_ElecEwSh_VdwLJEwSh_GeomW4W4_sse4_1_single.c 130KB nb_kernel_ElecEwSh_VdwLJEwSh_GeomW4W4_sse2_single.c 130KB nb_kernel_ElecEwSh_VdwLJEwSh_GeomW3W3_avx_256_double.c 129KB nb_kernel_ElecCSTab_VdwCSTab_GeomW4W4_sse2_single.c 128KB nb_kernel_ElecCSTab_VdwCSTab_GeomW4W4_sse4_1_single.c 128KB nb_kernel_ElecEw_VdwCSTab_GeomW4W4_avx_256_double.c 128KB nb_kernel_ElecCSTab_VdwCSTab_GeomW3W3_avx_256_double.c 125KB nb_kernel_ElecEw_VdwLJEw_GeomW4W4_avx_256_double.c 125KB nb_kernel_ElecEwSh_VdwLJSh_GeomW4W4_sse4_1_single.c 125KB nb_kernel_ElecEwSh_VdwLJSh_GeomW4W4_sse2_single.c 125KB nb_kernel_ElecCSTab_VdwLJ_GeomW4W4_avx_256_double.c 124KB nb_kernel_ElecEwSh_VdwLJEwSh_GeomW4W4_avx_256_single.c 124KB nb_kernel_ElecEwSh_VdwLJSh_GeomW3W3_avx_256_double.c 124KB nb_kernel_ElecCSTab_VdwCSTab_GeomW4W4_avx_128_fma_single.c 122KB nb_kernel_ElecCSTab_VdwCSTab_GeomW4W4_sse4_1_double.c 122KB nb_kernel_ElecEwSh_VdwLJEwSh_GeomW3W3_sse4_1_single.c 122KB nb_kernel_ElecEw_VdwCSTab_GeomW4W4_sse4_1_single.c 122KB nb_kernel_ElecEwSh_VdwLJEwSh_GeomW3W3_sse2_single.c 122KB nb_kernel_ElecCSTab_VdwCSTab_GeomW4W4_sse2_double.c 122KB nb_kernel_ElecEw_VdwCSTab_GeomW4W4_sse2_single.c 122KB nb_kernel_ElecEwSh_VdwLJEwSh_GeomW4W4_sse4_1_double.c 121KB nb_kernel_ElecEwSw_VdwLJSw_GeomW3W3_avx_128_fma_single.c 121KB nb_kernel_ElecEwSh_VdwLJEwSh_GeomW4W4_sse2_double.c 121KB nb_kernel_ElecEw_VdwCSTab_GeomW3W3_avx_256_double.c 121KB nb_kernel_ElecEw_VdwCSTab_GeomW4W4_avx_256_single.c 120KB nb_kernel_ElecEwSh_VdwLJEwSh_GeomW4W4_sparc64_hpc_ace_double.c 120KB nb_kernel_ElecEwSh_VdwNone_GeomW4W4_avx_256_double.c 120KB nb_kernel_ElecCSTab_VdwCSTab_GeomW3W3_sse2_single.c 120KB nb_kernel_ElecCSTab_VdwCSTab_GeomW3W3_sse4_1_single.c 120KB nb_kernel_ElecEwSh_VdwNone_GeomW3W3_avx_256_double.c 120KB nb_kernel_ElecEw_VdwLJ_GeomW4W4_avx_256_double.c 120KB nb_kernel_ElecEw_VdwLJEw_GeomW4W4_sse4_1_single.c 119KB nb_kernel_ElecEwSh_VdwLJEwSh_GeomW4W4_avx_128_fma_double.c 119KB nb_kernel_ElecEw_VdwLJEw_GeomW4W4_sse2_single.c 119KB nb_kernel_ElecCSTab_VdwLJ_GeomW4W4_sse2_single.c 119KB nb_kernel_ElecCSTab_VdwLJ_GeomW4W4_sse4_1_single.c 119KB nb_kernel_ElecCSTab_VdwCSTab_GeomW4W4_sparc64_hpc_ace_double.c 118KB nb_kernel_ElecEw_VdwLJEw_GeomW3W3_avx_256_double.c 118KB nb_kernel_ElecEwSh_VdwLJSh_GeomW4W4_avx_256_single.c 118KB nb_kernel_ElecCSTab_VdwLJ_GeomW3W3_avx_256_double.c 118KB gmx_bar.c 118KB nb_kernel_ElecEwSh_VdwLJSh_GeomW3W3_sse4_1_single.c 118KB nb_kernel_ElecEwSh_VdwLJSh_GeomW3W3_sse2_single.c 118KB nb_kernel_ElecCSTab_VdwCSTab_GeomW4W4_avx_128_fma_double.c 118KB nb_kernel_ElecEwSw_VdwNone_GeomW4W4_avx_128_fma_single.c 117KB nb_kernel_ElecEwSw_VdwNone_GeomW3W3_avx_128_fma_single.c 117KB nb_kernel_ElecEwSh_VdwLJSh_GeomW4W4_sse4_1_double.c 117KB nb_kernel_ElecEwSh_VdwLJSh_GeomW4W4_sse2_double.c 116KB
tng_io.c 648KB domdec.c 302KB nbnxn_search.c 187KB genborn_allvsall_sse2_single.c 181KB nb_kernel_ElecCSTab_VdwCSTab_GeomW4W4_avx_256_single.c 173KB pme.c 170KB nb_kernel_ElecEwSw_VdwLJSw_GeomW4W4_avx_256_double.c 168KB readir.c 164KB nb_kernel_ElecCSTab_VdwCSTab_GeomW3W3_avx_256_single.c 163KB bondfree.c 160KB nb_kernel_ElecEwSw_VdwLJSw_GeomW4W4_sse2_single.c 160KB nb_kernel_ElecEwSw_VdwLJSw_GeomW4W4_sse4_1_single.c 159KB nb_kernel_ElecEwSw_VdwLJSw_GeomW3W3_avx_256_double.c 158KB nb_kernel_ElecCSTab_VdwLJ_GeomW4W4_avx_256_single.c 157KB gmx_hbond.c 154KB nb_kernel_ElecEwSw_VdwNone_GeomW4W4_avx_256_double.c 154KB nb_kernel_ElecEwSw_VdwLJSw_GeomW4W4_sse4_1_double.c 154KB nb_kernel_ElecEwSw_VdwNone_GeomW3W3_avx_256_double.c 154KB nb_kernel_ElecEwSw_VdwLJSw_GeomW4W4_avx_256_single.c 154KB nb_kernel_ElecEwSw_VdwLJSw_GeomW4W4_sse2_double.c 154KB nb_kernel_ElecEwSw_VdwLJSw_GeomW4W4_sparc64_hpc_ace_double.c 152KB nb_kernel_ElecCSTab_VdwLJ_GeomW3W3_avx_256_single.c 150KB nb_kernel_ElecEwSw_VdwLJSw_GeomW3W3_sse4_1_single.c 150KB nb_kernel_ElecEwSw_VdwLJSw_GeomW3W3_sse2_single.c 150KB nb_kernel_ElecEwSw_VdwLJSw_GeomW4W4_avx_128_fma_double.c 150KB nb_kernel_ElecCSTab_VdwNone_GeomW4W4_avx_256_single.c 147KB nb_kernel_ElecCSTab_VdwNone_GeomW3W3_avx_256_single.c 147KB nb_kernel_ElecEwSw_VdwNone_GeomW4W4_sse4_1_single.c 146KB nb_kernel_ElecEwSw_VdwNone_GeomW4W4_sse2_single.c 146KB nb_kernel_ElecEwSw_VdwNone_GeomW3W3_sse4_1_single.c 146KB nb_kernel_ElecEwSw_VdwNone_GeomW3W3_sse2_single.c 146KB nb_kernel_ElecEwSw_VdwLJSw_GeomW3W3_sse4_1_double.c 145KB nb_kernel_ElecEwSw_VdwLJSw_GeomW3W3_sse2_double.c 144KB nb_kernel_ElecEwSw_VdwLJSw_GeomW3W3_avx_256_single.c 143KB nb_kernel_ElecEwSw_VdwLJSw_GeomW3W3_sparc64_hpc_ace_double.c 143KB pull_rotation.c 142KB nb_kernel_ElecEwSw_VdwLJSw_GeomW3W3_avx_128_fma_double.c 141KB nb_kernel_ElecEwSw_VdwNone_GeomW4W4_sse4_1_double.c 141KB nb_kernel_ElecEwSw_VdwNone_GeomW3W3_sse4_1_double.c 141KB nb_kernel_ElecEwSw_VdwNone_GeomW4W4_sse2_double.c 140KB nb_kernel_ElecEwSw_VdwNone_GeomW3W3_sse2_double.c 140KB nb_kernel_ElecEwSw_VdwNone_GeomW4W4_avx_256_single.c 140KB nb_kernel_ElecEwSw_VdwNone_GeomW3W3_avx_256_single.c 139KB nb_kernel_ElecEwSw_VdwNone_GeomW4W4_sparc64_hpc_ace_double.c 139KB nb_kernel_ElecEwSw_VdwNone_GeomW3W3_sparc64_hpc_ace_double.c 139KB nb_kernel_ElecEwSw_VdwNone_GeomW4W4_avx_128_fma_double.c 137KB nb_kernel_ElecEwSw_VdwNone_GeomW3W3_avx_128_fma_double.c 137KB nb_kernel_ElecEwSh_VdwLJEwSh_GeomW4W4_avx_256_double.c 137KB nb_kernel_ElecCSTab_VdwCSTab_GeomW4W4_avx_256_double.c 134KB gmx_arpack.c 133KB nb_kernel_ElecEwSh_VdwLJSh_GeomW4W4_avx_256_double.c 131KB nb_kernel_ElecEwSw_VdwLJSw_GeomW4W4_avx_128_fma_single.c 130KB nb_kernel_ElecEwSh_VdwLJEwSh_GeomW4W4_sse4_1_single.c 130KB nb_kernel_ElecEwSh_VdwLJEwSh_GeomW4W4_sse2_single.c 130KB nb_kernel_ElecEwSh_VdwLJEwSh_GeomW3W3_avx_256_double.c 129KB nb_kernel_ElecCSTab_VdwCSTab_GeomW4W4_sse2_single.c 128KB nb_kernel_ElecCSTab_VdwCSTab_GeomW4W4_sse4_1_single.c 128KB nb_kernel_ElecEw_VdwCSTab_GeomW4W4_avx_256_double.c 128KB nb_kernel_ElecCSTab_VdwCSTab_GeomW3W3_avx_256_double.c 125KB nb_kernel_ElecEw_VdwLJEw_GeomW4W4_avx_256_double.c 125KB nb_kernel_ElecEwSh_VdwLJSh_GeomW4W4_sse4_1_single.c 125KB nb_kernel_ElecEwSh_VdwLJSh_GeomW4W4_sse2_single.c 125KB nb_kernel_ElecCSTab_VdwLJ_GeomW4W4_avx_256_double.c 124KB nb_kernel_ElecEwSh_VdwLJEwSh_GeomW4W4_avx_256_single.c 124KB nb_kernel_ElecEwSh_VdwLJSh_GeomW3W3_avx_256_double.c 124KB nb_kernel_ElecCSTab_VdwCSTab_GeomW4W4_avx_128_fma_single.c 122KB nb_kernel_ElecCSTab_VdwCSTab_GeomW4W4_sse4_1_double.c 122KB nb_kernel_ElecEwSh_VdwLJEwSh_GeomW3W3_sse4_1_single.c 122KB nb_kernel_ElecEw_VdwCSTab_GeomW4W4_sse4_1_single.c 122KB nb_kernel_ElecEwSh_VdwLJEwSh_GeomW3W3_sse2_single.c 122KB nb_kernel_ElecCSTab_VdwCSTab_GeomW4W4_sse2_double.c 122KB nb_kernel_ElecEw_VdwCSTab_GeomW4W4_sse2_single.c 122KB nb_kernel_ElecEwSh_VdwLJEwSh_GeomW4W4_sse4_1_double.c 121KB nb_kernel_ElecEwSw_VdwLJSw_GeomW3W3_avx_128_fma_single.c 121KB nb_kernel_ElecEwSh_VdwLJEwSh_GeomW4W4_sse2_double.c 121KB nb_kernel_ElecEw_VdwCSTab_GeomW3W3_avx_256_double.c 121KB nb_kernel_ElecEw_VdwCSTab_GeomW4W4_avx_256_single.c 120KB nb_kernel_ElecEwSh_VdwLJEwSh_GeomW4W4_sparc64_hpc_ace_double.c 120KB nb_kernel_ElecEwSh_VdwNone_GeomW4W4_avx_256_double.c 120KB nb_kernel_ElecCSTab_VdwCSTab_GeomW3W3_sse2_single.c 120KB nb_kernel_ElecCSTab_VdwCSTab_GeomW3W3_sse4_1_single.c 120KB nb_kernel_ElecEwSh_VdwNone_GeomW3W3_avx_256_double.c 120KB nb_kernel_ElecEw_VdwLJ_GeomW4W4_avx_256_double.c 120KB nb_kernel_ElecEw_VdwLJEw_GeomW4W4_sse4_1_single.c 119KB nb_kernel_ElecEwSh_VdwLJEwSh_GeomW4W4_avx_128_fma_double.c 119KB nb_kernel_ElecEw_VdwLJEw_GeomW4W4_sse2_single.c 119KB nb_kernel_ElecCSTab_VdwLJ_GeomW4W4_sse2_single.c 119KB nb_kernel_ElecCSTab_VdwLJ_GeomW4W4_sse4_1_single.c 119KB nb_kernel_ElecCSTab_VdwCSTab_GeomW4W4_sparc64_hpc_ace_double.c 118KB nb_kernel_ElecEw_VdwLJEw_GeomW3W3_avx_256_double.c 118KB nb_kernel_ElecEwSh_VdwLJSh_GeomW4W4_avx_256_single.c 118KB nb_kernel_ElecCSTab_VdwLJ_GeomW3W3_avx_256_double.c 118KB gmx_bar.c 118KB nb_kernel_ElecEwSh_VdwLJSh_GeomW3W3_sse4_1_single.c 118KB nb_kernel_ElecEwSh_VdwLJSh_GeomW3W3_sse2_single.c 118KB nb_kernel_ElecCSTab_VdwCSTab_GeomW4W4_avx_128_fma_double.c 118KB nb_kernel_ElecEwSw_VdwNone_GeomW4W4_avx_128_fma_single.c 117KB nb_kernel_ElecEwSw_VdwNone_GeomW3W3_avx_128_fma_single.c 117KB nb_kernel_ElecEwSh_VdwLJSh_GeomW4W4_sse4_1_double.c 117KB nb_kernel_ElecEwSh_VdwLJSh_GeomW4W4_sse2_double.c 116KB












评论0