MS分子动力学模拟具体实施步骤
需积分: 48 192 浏览量
2019-01-26
10:31:29
上传
评论 18
收藏 683KB PDF 举报

第 3 章 铁基块体非晶合金‐纳米晶转变的动力学模拟过程
3.1Discover 模块
3.1.1 原子力场的分配
在使用 Discover 模块建立基于力场的计算中,涉及几个步骤。主要有:选择
力场、指定原子类型、计算或指定电荷、选择 non‐bondcutoffs。
在这些步骤中,指定原子类型和计算电荷一般是自动执行的。然而,在某些
情形下需要手动指定原子类型。原子定型使用预定义的规则对结构中的每个原子
指定原子类型。在为特定的系统确定能量和力时,定型原子使工作者能使用正确
的力场参数。通常,原子定型由 Discover 使用定型引擎的基本规则来自动执行,
所以不需要手动原子定型。然而,在特殊情形下,人们不得不手动的定型原子,
以确保它们被正确地设置。
图 3-1
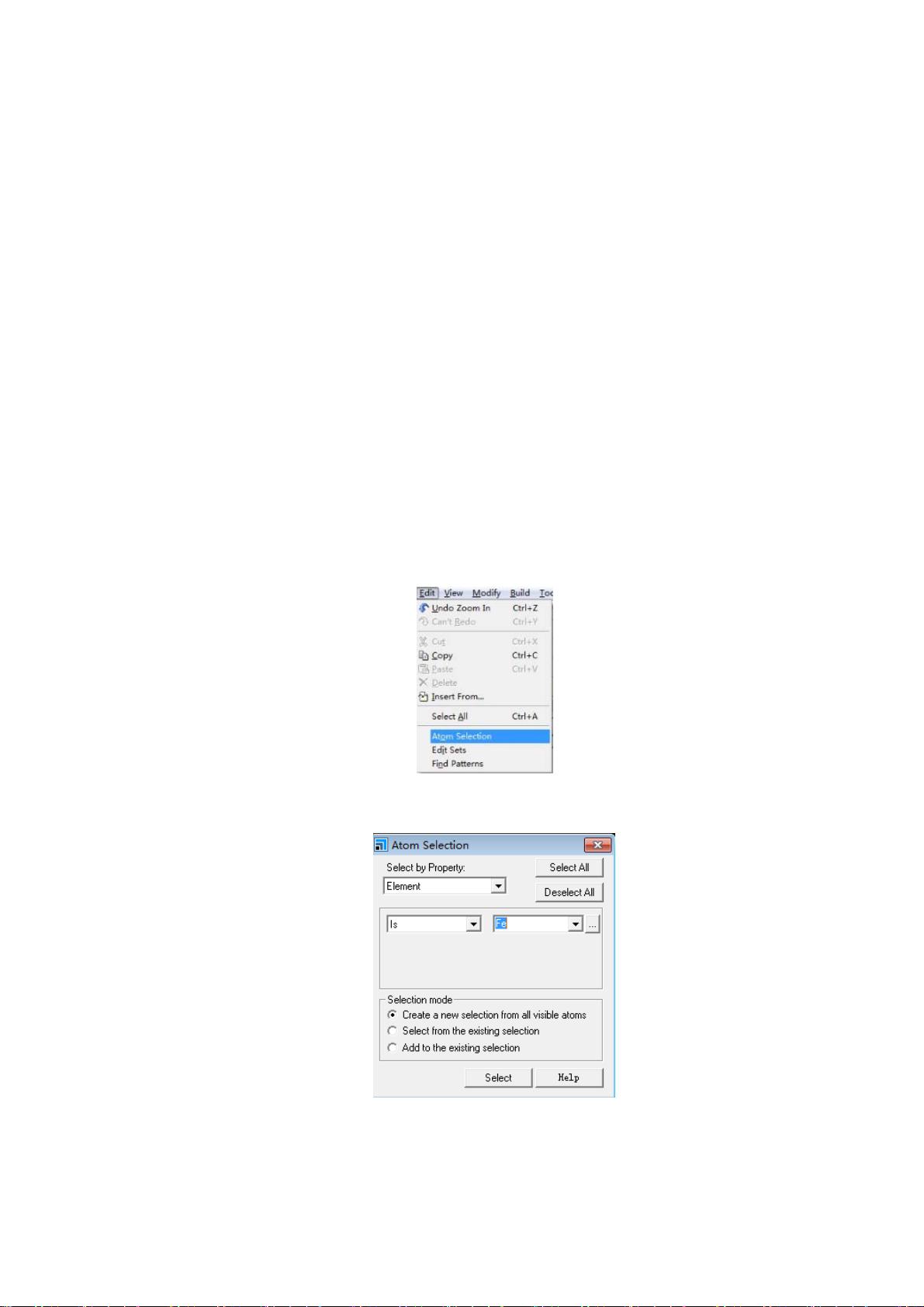
1) 计算并显示原子类型:点击 Edit→AtomSelection ,如图 3‐1 所示
图3-2
弹出对话框,如图 3‐2 所示
从右边的…的元素周期表中选择 Fe,再点 Select,此时所建晶胞中所有 Fe
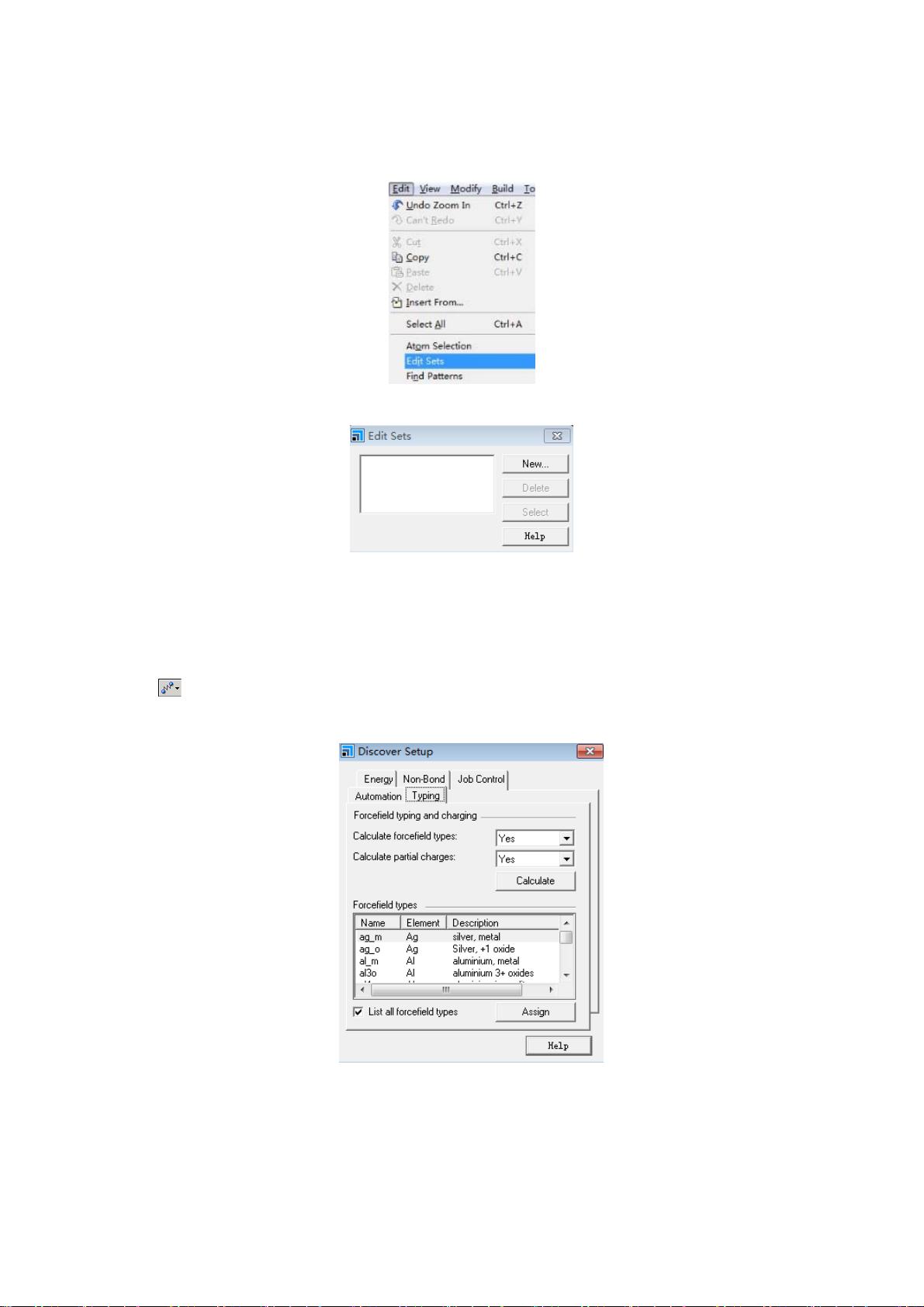
剩余20页未读,继续阅读
资源评论


yin29922990
- 粉丝: 2
- 资源: 1









最新资源
- 转载使用许可协议范本(互联网行业)模版.doc
- 软件产业运行情况调研问卷模版.doc
- 软件产品发布管理流程.doc
- 软件仿真多机串行通信.doc
- Python大作业:音乐播放软件(爬虫+可视化+数据分析+数据库)
- 课程设计-python爬虫-爬取日报,爬取日报文章后存储到本地,附带源代码+课程设计报告
- 软件和信息技术服务行业投资与前景预测.pptx
- 课程设计-基于SpringBoot + Mybatis+python爬虫NBA球员数据爬取可视化+源代码+文档+sql+效果图
- 软件品质管理系列二项目策划规范.doc
- 基于TensorFlow+PyQt+GUI的酒店评论情感分析,支持分析本地数据文件和网络爬取数据分析+源代码+文档说明+安装教程
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


